VİTREOMAKULER ARA YÜZEY HASTALIKLARI
Vitreomakuler Traksiyon ve Makula Deliği
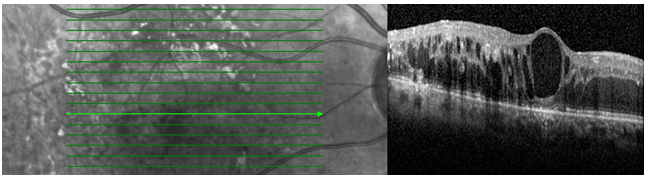
Arka vitreusun yaşa bağlı dekolmanı doğal bir süreç olmakla birlikte, bazen vitreus korteksi ve makula arasındaki vitreomakuler adezyonların (VMA) sıkı olması nedeniyle komplike olabilir. Bu adezyonlar dar ya da geniş olabilir, sadece foveolayı ya da daha geniş bir makuler alanı etkileyebilir (Resim 2). Basit asemptomatik VMA’larda makulanın anatomik yapısı korunur. Ancak VMA’lar makulaya ön-arka yönde çekme gücü oluşturduğunda retinada yapısal değişikliğe neden olur, bu tablo vitreomakuler traksiyon (VMT) olarak tanımlanır (Resim 3). VMT intraretinal kist oluşumu ve retina dekolmanına neden olabilir. VMA’nın genişliği arttıkça retinaya uyguladığı traksiyon gücü ve foveada oluşturduğu deformasyon azalır (1).
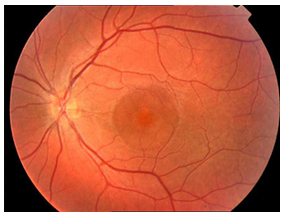
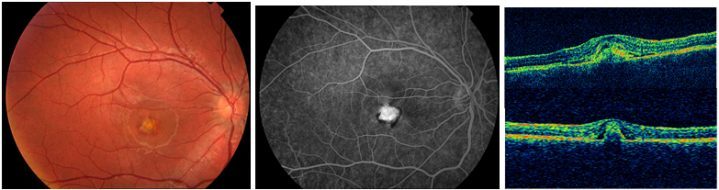
Makula deliği (MD), foveal nörosensöryel retinada yuvarlak tam kat retina defektidir (Resim 1). Travma, miyopi veya enflamasyona bağlı geliştiği bildirilmiş olsa da büyük oranda idiyopatiktir (2). İdiyopatik makula deliği (İMD) 6 ve 7. dekatta, kadınlarda sık görülür. %80 tek taraflıdır (3).
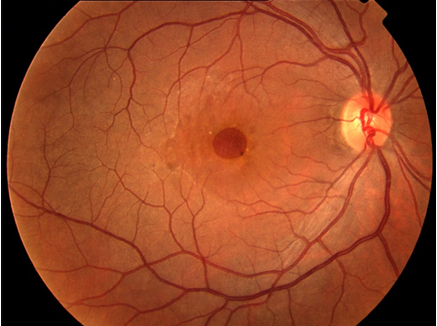
Resim 1: İMD olan hastanın renkli fundus fotoğrafı. Büyük, tam kat makula deliği izlenmekte.
Patogenez
Gass, İMD patogenezinde, prefoveal vitreus korteksinde tanjansiyel traksiyonun foveada dekolman ve tam kat MD gelişimi ile sonuçlandığını savunmuştur (4). Son çalışmalar MD gelişiminde, ön-arka ve dinamik vitreomakuler traksiyon sonucu perifoveal arka vitreus dekolmanının etkili olduğunu göstermiştir (5).
Evreleme
Gass makula deliklerini klinik muayeneyi temel alarak 4 evre olarak sınıflamıştır. Gass sınıflamasının adaptasyonları halen klinik kullanımda olsa da, optik koherens tomografi (OKT) ile sağlanan anatomik bilgi makula deliğinin patogenezinin daha iyi anlaşılmasına katkıda bulunmuştur. 2013 Eylül de Uluslararası Vitreomakuler Traksiyon Çalışma Grubu OKT’ye dayanarak yeni bir Vitreomakuler adezyon, traksiyon ve makula deliği sınıflaması yayınlamıştır ve artık bu sınıflamaya göre terminoloji kullanılmaktadır (Tablo 1, Tablo 2) (Resim 2,3,4)(6).
Gass Sınıflaması (7)
Evre 1a: Delik tehdidi; foveanın düzleşmesi sonucu foveal çöküntü kaybolur, sarı nokta görülür. İç retina tabakaları fotoreseptör tabakadan ayrılarak skizis kavitesi oluşturur. Yeni sınıflamada “intrafoveal psödokistli fokal VMT” olarak adlandırılan tabloya karşılık gelmektedir (Resim 3C).
Evre 1b: Gizli makula deliği; foveada sarı halka görünümü izlenir. Arka hyaloid foveaya tutunarak , foveolar bölgede sensöryel retina ve retina pigment epiteli tabakalarını ayırır, retina içi kistik değişiklikler oluşur.
Evre 2: Küçük tam kat makula deliği; makula deliğinin çapı 400 µm’dan küçüktür. Delik merkezi, hafif merkezin dışında yerleşmiş ya da hilal şeklinde olabilir. Vitreofoveolar yapışıklık korunduğu halde sikizis kavitesinin tavanı ayrılır. Yeni sınıflamada VMT’nin devam ettiği orta büyüklükte TKMD (Resim 4B).
Evre 3: Tam kat makula deliği; makula deliğinin çapı 400 µm’dan büyüktür. Skizis kavitesinin çatısı ayrılır ve operkulum oluşur. Vitreusun parafoveal yapışıklığı korunmaktadır. Makula deliğinin tabanı kırmızıdır. Subretinal sıvı birikimine bağlı olarak deliği çevreleyen gri bir halka ve deliğin üzerinde operkulum klinik olarak izlenebilir. Yeni sınıflamada VMT’siz tam kat büyük makula deliği (Resim 4C).
Evre 4: Tam kat makula deliği ve total arka vitreus dekolmanı (AVD); Weiss halkası izlenir (7). Yeni sınıflamada VMT’siz tam kat büyük makula deliği (Resim 4C).
Tablo 1: Uluslararası Vitreomakuler Traksiyon Çalışma Gurubunun Vitreomakuler Adezyon, Traksiyon Ve Makula Deliği Sınıflaması
| Sınıflama | Alt sınıflama |
| Vitreomakuler adezyon (VMA) | Boyut: dar (≤1500µm) ya da geniş (>1500 µm) |
| Tek başına ya da ek patoloji eşliğinde | |
| Vitreomakuler traksiyon (VMT) | Boyut: dar (≤1500µm) ya da geniş (>1500 µm) |
| Tek başına ya da ek patoloji eşliğinde | |
| Tam kat makula deliği (TKMD) | Boyut: küçük (≤250µm), orta (>250-≤400µm),büyük (>400µm) |
| Vitreusun durumu: VMT var veya yok | |
| Etyoloji: primer veya sekonder | |
| Lamellar Delik | İç foveal defekt, intraretinal skizis, fotoreseptör tabaka intakt. |
| Psödohol | Foveal ERM (foveola hariç), santral fovea normal, foveal kontür dik, biyomikroskopta delik benzeri görünüm. |
Resim 2: (A) Dar VMA ≤1500µm, (B) geniş VMA >1500µm
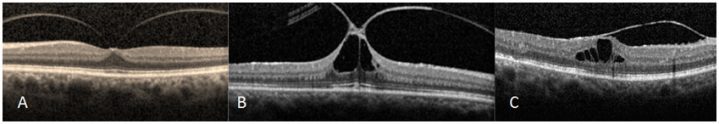
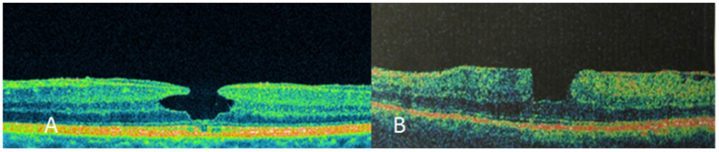
Resim 3: (A) Dar VMT ≤1500µm, (B) Dar VMT foveada traksiyon ve kist oluşumu, (C) Geniş VMT >1500µm, foveada intraretinal kistoid değişiklikler izlenmekte.
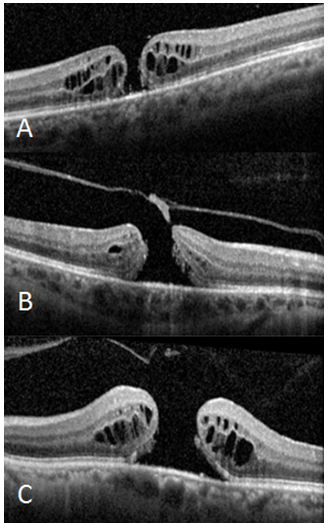
Resim 4: (A) Küçük TKMD, VMT yok. Foveal lezyon internal limitan membrandan retina pigment epiteline kadar bütün katları içeriyor, lezyon genişliği ≤250µm. (B) Orta TKMD ve VMT. Deliğin genişliği 250-400µm, Vitreus delik kenarına tutunmuş. (C)Büyük TKMD, VMT yok. Deliğin genişliği >400µm, arka vitreus total dekole, retinada kistoid değişiklikler.
Lamellar delik ile psödohol birbiri ile sıklıkla karıştırılabilen iki antitedir. Lamellar delikte tipik olarak iç retinal katlardaki defektin kenarlarından skizis şeklinde retina içine uzanan ayrılmalar görülür ve fovea genel olarak incelmiştir (Resim 5A).
Psödoholde ise mutlaka beraberinde ERM eşlik eder ve bu ERM’nin fovea merkezindeki bölümünde açıklık vardır. Fovea kenar kontürü dikleşmiştir ve yumuşak kavsi bozulmuştur, foveal kalınlık genellikle artmıştır ve retina katlarına uzanan skizis şeklinde ayrılmalar görülmez (Resim 5B).
Resim 5: (A) Lameller makula deliği, (B) Psödohol
Tablo 2: Gass’ın Makula Deliği Evrelemesi ve Uluslararası Vitreomaküler Traksiyon Çalışma Gurubunun Vitreomakuler Adezyon, Traksiyon Ve Makula Deliği Sınıflamasının Korelasyonu
| Makula Deliği Gass Sınıflaması | Uluslararası Vitreomakuler Traksiyon Çalışma Gurubunun Sınıflaması |
| Evre 0 | VMA |
| Evre 1 : delik tehdidi | VMT |
| Evre 2: küçük delik | Küçük, orta TKMD + VMT |
| Evre 3: büyük delik | Orta, büyük TKMD + VMT yok |
| Evre 4: TKMD ve AVD | Küçük, orta, büyük TKMD, VMT yok |
Klinik
VMT erişkin yaşın hastalığıdır, görme keskinliğinde azalma, metamorfopsi, fotopsi ve mikropsiye neden olabilir.
VMT ve VMT eşliğindeki küçük, orta TKMD genellikle asemptomatiktir, özellikle diğer göz sağlıklı ve dominant olmayan göz tutulmuş ise hastanın görme ile ilgili bir şikayeti bulunmaz. İleri evre makula deliklerinde (evre 3-4) ise görme belirgin olarak azalır, metamorfopsi, merkezi görme kaybı ve merkezi skotom gelişimi izlenir. Görme keskinliği makula deliğinin büyüklüğü ile ters orantılıdır (1). Psödoholler genellikle tesadüfen farkedilir. Hastanın hiç şikayeti yokken klinik görünümü çok dramatik olabilir.
Tanı Yöntemleri
1.Watzke-Allen Testi: Biyomikroskobun ışığı dar bir yarık şeklinde vertikal olarak foveaya yönlendirilir. Makula deliği varlığında ışığın kırık- iki parça şeklinde veya incelmiş görüntüsü beklenir. Yalancı delik, lamellar delik ya da kist varlığında şekli düzensiz tek ışık görüntüsü izlenir.
2.Optik Koherens Tomografi (OKT): Vitreoretinal yüzel hastalıkları hakkındaki bilgimiz OKT görüntüleme yönteminin kullanıma girmesi ile artmıştır. OKT vitreoretinal anatominin girişimsel olmayan bir yöntemle yüksek çözünürlükte görüntüler alınarak detaylı analizine imkan sağlamakta böylece vitreoretinal yüzeyin patolojileri daha net ve doğru olarak tanımlanmaktadır. OKT makula deliğinin tanısı, evrelemesi, tedavinin planlanması ve takibinde vazgeçilmez bir görüntüleme yöntemidir . Son yıllarda Spektral domain OKT (SD-OKT)’nin yaygın kullanımı, vitreoretinal ara yüzey ve retinanın daha yüksek çözünürlükte görüntülerinin alınmasına imkan vermiştir. SD-OKT makula deliğinin taban genişliği, çapı, yüksekliği gibi parametrelerinin ölçülmesinde faydalıdır. Ayrıca İMD, makuler psödohol ya da lameller makula deliği ayırıcı tanısında kullanılır (8).
- Floresein anjiografi (FA): Tam kat makula deliği varlığında ksantofil pigmentinin yer değiştirmesi ve RPE atrofisine bağlı erken pencere defekti izlenir. Geç dönem görüntülerinde deliği çevreleyen subretinal sıvı nedeniyle hiperfloresan halka görünümü bulunabilir.
Tedavi
Dar VMA’lar kendiliğinden gerileyebildiğinden, asemptomatik hastalarda amsler grid testi ile takip önerilmektedir. Metamorfopsi ve skotom varlığında rutin olarak OKT ile görüntüleme yapılmalıdır.
VMT kendiliğinden gerileyebilir. Vitreusun spontan ayrılması ile traksiyon rahatlayarak intraretinal psödokist görünümü yok olabilir ve semptomlar gerileyebilir. Diğer taraftan traksiyon devam eder ve spontan ayrılma gelişmezse tam kat MD otraya çıkabilir. Yakın takip ile konservatif davranılmalı ve görmede belirgin azalmaya neden olan durumlarda cerrahi düşünülmelidir.
Cerrahi Tedavi
1991’de Kelly ve Wendel makula deliği tedavisinde vitrektomi ve göz içi gaz tamponadı prosedürünü tanımlamıştır. Çalışmaya dahil edilen 52 hastanın çoğunluğunda evre 3 MD mevcutken, cerrahi ile deliğin kapanma oranı %58 olarak bildirilmiş, 6 ay sonra %73 oranında 2 sıra ve üstünde görme keskinliği artışı saptanmıştır (9). Günümüzde, MD’nin operasyon öncesi boyutları, semptomların süresi, kullanılan cerrahi tekniğe bağlı olarak cerrahi ile %90’ın üstünde anatomik başarı elde edilebilmektedir.
TKMD olgularında görme 6/9’un altındaysa cerrahi önerilmektedir. VMT’nin eşlik ettiği küçük, orta delik (<400µm), 6 aydan az süredir semptomatik olan delikler iyi prognoz göstergesidir (10).
Uygulanan cerrahinin hedefi, tanjansiyel ve ön-arka vitreus traksiyonunun ortadan kaldırılması, vitrektomi ve epiretinal membran (ERM) soyulması ile delik kenarındaki glial hücrelerin aktive edilmesi, göz içi gaz tamponadı ve yüz üstü pozisyon vererek deliğin kenarlarının apozisyonunun sağlanmasıdır. Son dekatta, gliozisi indüklemek, retinal elastikiyeti arttırmak ve anatomik başarıyı artırmak amacıyla internal limitan membranın (İLM) soyulması tekniği yaygınlık kazanmıştır (11-13). ILM boyasız olarak da soyulabileceği gibi, indosiyanin yeşili, tripan mavisi, brilliant mavisi boyaları ve triamsinolon ile de boyamak suretiyle görünürlüğünü artırarak daha non-travmatik bir şekilde soyulması mümkündür. Kliniğimizde bu amaçla en çok brillant mavisi kullanılmaktadır (Resim 6).
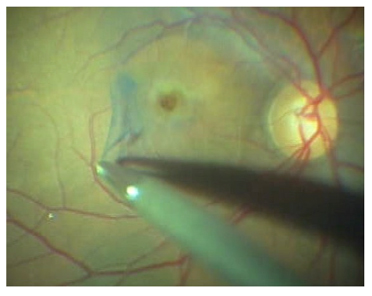
Resim 6: Bir makula deliği cerrahisinde Brillant mavisi ile ILM soyulması.
Vitrektomi ve İLM soyulması tamamlandıktan sonra perifer retina yırtık varlığını ekarte etmek için kontrol edilir, ve hava-sıvı değişimini takiben göz içine gaz verilir. Bu amaçla sıklıkla kullanılan gazlar perfloropropan (C3F8) ve sülfür hekzaflorid (SF6) dir. Göz içine verilen gaz iki mekanizma ile makula deliğinin kapanmasına aracılık eder; 1) delik üzerindeki gazın yüzey gerilimi vitreus boşluğundan deliğe doğru sıvı akımını engeller, 2) gaz yüzeyi makula deliğinin kenarları arasında bir köprü görevi görerek glial hücrelerin göçüne katkıda bulunur (14). Tampon madde olarak silikon yağı kullanımı da mümkündür. Silikon genellikle uçak yolculuğu zorunlu olan hastalarda gaz kullanılamayacağından gündeme gelir. Özellikle yüzüstü pozisyon sağlanamayan hastalarda ağır silikon da düşünülebilir. Diğer taraftan son dönemlerde tampon olarak sadece hava verilip 2-3 gün yüzüstü pozisyon verilmesinin de yeterli olabileceği konusunda da görüşler vardır (15) (Resim 7).
Cerrahi sonrası yüzüstü pozisyonu önerilir. Ancak özellikle küçük deliklerde bunun her zaman gerekli olmadığı düşünülmektedir. Hasta bütün süre boyunca supin pozisyonda yatmadığı sürece gaz makula deliği yüzeyindeki köprü görevini yerine getirebilir.
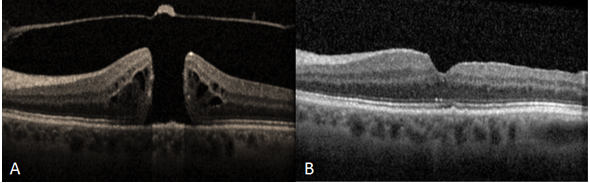
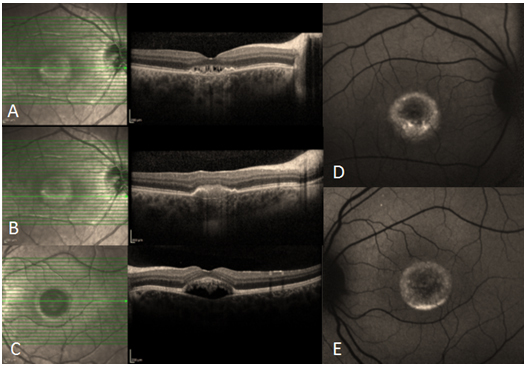
Resim 7: (A) 510µm genişliğinde TKMD olan hastanın preoperatif OKT görüntüsü, VMT foveadan serbestleşmiş, operkülüm izlenmekte, delik kenarlarında retina içi kistler ve kalınlaşma mevcut, görme keskinliği (GK) 0.1. (B) vitrektomi + internal limitan membran soyulması + SF6 gaz tamponadı + yüz üstü poziyon verilmesi ameliyatı sonrası 1. Ay OKT görüntüsü, foveal kontürün tekrar oluştuğu görülmekte, GK 0.6.
Farmakolojik vitreoliz
Farmakolojik vitreolizis vitreus yapısının kimyasal olarak değiştirilmesi yolu ile vitreusu sıvılaştırıp, vitreoretinal bağlantıları zayıflatarak güvenli bir AVD’nı hedefleyen gelişmekte olan bir medikal tedavi yöntemidir. İntravitreal okriplazmin enjeksiyonu semptomatik vitreomaküler traksiyonlu olgularda ve VMT’li küçük-orta (<400µm) makula deliklerinde cerrahiye alternatif olarak gündeme gelmiş yeni bir yöntemdir (16,17). Bu olgularda tek enjeksiyonla 1. Ayın sonunda %40 civarında delikte kapanma bildirilmektedir. Plaseboda bu oran %10 civarındadır.
İMD semptomatik ise ve hastanın hayat kalitesini etkiliyor ise erken dönemde tedavi edilmelidir. Makula deliğinin çapı <400µm ise ve vitreomaküler traksiyon (VMT) mevcut ise Farmakolojik vitreoliz bir tedavi seçeneği olarak düşünülebilir. Hastanın sistemik morbiditesi ve cerrahiye uygunluğu gözönünde bulundurulmalıdır. Eğer farmakolojik vitreolitik ajanın enjeksiyonunu takiben 1 ay içinde delik kapanmıyorsa vitrektomi yapılmalıdır. İMD’nin çapı >400µm ise, beraberinde ERM mevcutsa ve VMT yoksa ilk tedavi seçeneği vitrektomi olmalıdır (1).
Epiretinal Maküler Membranlar
Epiretinal membranlar (ERM), retina yüzeyinde fibroselüler proliferasyona bağlı oluşan membranlardır. Bu klinik görünüm, preretinal makuler fibrozis, epiretinal fibrozis ya da gliozis, makuler pucker, yüzey kırışıklık retinopatisi, veya selofan makulopati olarak ta isimlendirilmiştir. Genellikle 50 yaş ve üstünün hastalığıdır (18).
ERM; en sık idyopatik olarak izlenir ancak ikincil nedenleri arasında diabetik retinopati ve retina ven tıkanıklığı gibi retina damar hastalıkları, retina dekolmanı cerrahisi, retina yırtıkları, laser fotokoagülasyon, kriyoterapi, travma ve enflamasyon bulunmaktadır.
ERM, hafif vakalarda asemptomatik olabilir ancak, membran kalınlığına ve retinada yaptığı şekil bozukluğuna bağlı olarak metamorfopsi ve görme keskinliğinde azalmaya neden olur (19).
Gass, ERM’ları klinik görünümüne göre evrelendirmiştir (20).
Evre 0: Selofan makulopati; makulada saydam bir membran vardır ve retinada şekil bozukluğuna yol açmaz. Muayenede retina yüzeyinde şeffaf ışık yansıması izlenir, asemptomatiktir.
Evre 1: Kırışık selofan makulopati; hastalık ilerlemiştir, ERM’deki büzüşme ve kontraksiyon iç retina katlarında kırışıklığa yol açar. Makula kapillerlerinde kıvrımlaşma görülebilir. İç retina katlarındaki bu şekil bozukluğu görme keskinliğinde düşmeye neden olur. Fovea etkilendiyse hastalar çarpık görme ve merkezi görmede bulanıklıktan şikayet ederler.
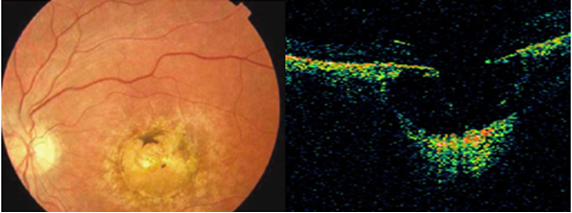
Evre 2: Preretinal makuler fibrozis; daha kalın ve opak bir membran izlenir. Alttaki retina damarlarını gizleyebilir ve retinanın tüm katlarını etkileyen belirgin şekil bozukluğuna yol açar (Resim 8). Retinada ödem, küçük hemorajiler, cotton-wool spotlar ve eksudalar beraberinde izlenebilir. “Makuler pucker” olarak da adlandırılır. Hastaların %80’i bulanık görme ve metamorfopsiden şikayet eder.
Tanı Testleri
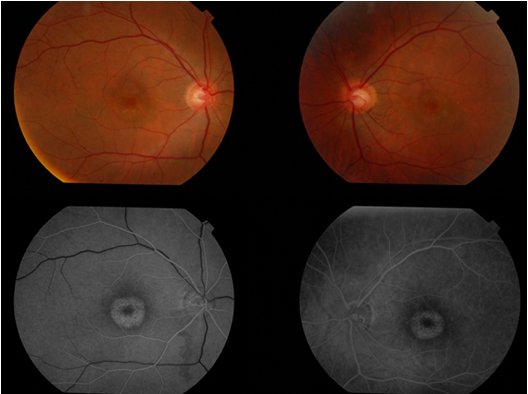
1.Optik Koherens Tomografi (OKT): OKT tanı koymada çok yararlıdır. Görüntülerde retina yüzeyinde yüksek yansıtıcılık gösteren (beyaz) tabaka izlenir, ayrıca retinada yaptığı kalınlaşma, katlantı, psödohol, makula ödemi gibi şekil bozuklukları da izlenir (Resim 9).
2.Floresein anjiografi (FA): İleri evre ERM’lerde, ağır maküler distorsiyona bağlı retina ödemi ve kan-retina bariyerinde bozulmaya bağlı sızıntı görülebilir. ERM sekonder ise özellikle retinal vasküler veya enflamatuar hastalıklara ait ipuçları görülebilir.
Tedavi
Rahatsız edici düzeyde metamorfopsi varlığında, görme keskinliği 0.5’in altına düşmüşse, binoküler görme etkilenmişse, semptom ve bulgularda ilerleme varsa, hasta durumundan şikayetçi ise tedavi cerrahidir (Resim 10) . Pars plana vitrektomi ve ERM soyulması standart yöntemdir. ERM ile beraber iç limitan membran (ILM) soyulması residüel membran kalmadığından emin olunmasını sağlayarak başarıyı arttırmakta ve özellikle yeniden membran oluşumunu engellemektedir (18).
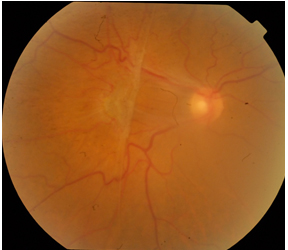
Resim 8: Sağ gözde makuler pucker olan hastanın renkli fundus fotoğrafı, makulada kalın ve opak membran, alttaki damarlarda ve retinada belirgin şekil bozukluğu izlenmekte.
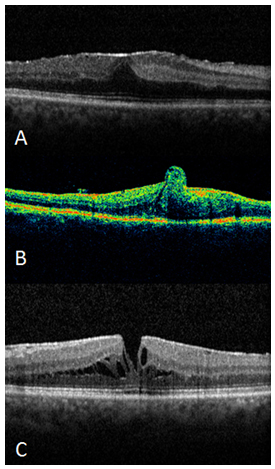
Resim 9: ERM, (A) foveal kontür düzleşmiş, (B) foveada retinal katlantı, (C) psödohol
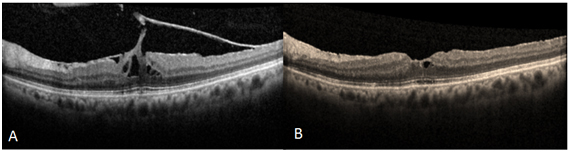
Resim 10: (A) ERM ve VMT’si olan hastanın cerrahi öncesi OKT görüntüsü, görme keskinliği 0.3 düzeyinde. (B) Cerrahi sonrası 1. Ay OKT görüntüsü. Görme keskinliği 0.6 düzeyinde.
KİSTOİD MAKULA ÖDEMİ
Makula ödemi, makulada dış pleksiform tabaka ve iç nükleer tabakada sıvı birikimi ve retinada kalınlaşma ile karakterizedir. Kistik alanlarla karakterize ise kistoid makula ödemi (KMÖ) olarak adlandırılır. KMÖ’de klasik görünüm kistlerin dış pleksiform tabakada bulunması olsa da, altta yatan nedene bağlı olarak retinanın diğer katları da tutulabilir (21).
Patogenez
Sağlıklı bir gözde, nörosensöriyel retinanın hücre dışı alandaki sıvı içeriği, retina kapiller endotel hücreleri arasındaki sıkı bağlantılar (iç kan retina bariyeri), retina pigment epiteli hücreleri arasındaki sıkı bağlantılar (dış kan retina bariyeri) ve RPE hücrelerinin pompa fonksiyonu tarafından düzenlenmektedir. İç ve dış kan retina bariyerini veya RPE’nin pompa fonksiyonunu bozan faktörler retinada sıvı birikimine neden olurlar. Artmış damar geçirgenliği, artmış kan akımı, RPE bariyer ve pompa fonksiyon bozukluğu, traksiyon yapan nedenler, ilaçlar patogenezde rol oynar (21).
Klinik
Hastaların en önemli yakınması görme azlığıdır. Ayrıca metamorfopsi, mikropsi, skotom ve fotofobi eşlik edebilir (22).
KMÖ Nedenleri
Göz içi cerrahi: Katarakt cerrahisini takiben gelişen KMÖ (Irvine-Gass Sendromu), cerrahi geçiren hastaların %1-2’sinde görme keskinliğinde azalmaya neden olur. Cerrahi sırasında arka kapsül açılması, vitreus kaybı, yara yerine vitreus adezyonu, iris travması, lens bakiyesi kalması gibi cerrahi komplikasyonlar KMÖ gelişimini artıran faktörlerdir (21).
Diğer KMÖ nedenleri aşağıda sıralanmıştır:
Lazer tedavileri: Panretinal lazer fotokoagülasyon, Nd:YAG lazer kapsülotomi, lazer iridotomi.
Üveitler: Pars planit, birdshot koryoretiniti, sarkoidoz, Behçet hastalığı, Vogt-Koyanagi-Harada hastalığı, toksoplazmozis, CMV retiniti, idiopatik vitritis.
Retinanın vasküler hastalıkları: Diyabetik retinopati, radyasyon retinopatisi, retina ven tıkanıklığı, hipertansif retinopati, retinal arter makroanevrizması, idyopatik jukstafoveal telenjektazi ve Coats hastalığı.
Koroidal vasküler hastalıklar: Koroid neovaskülarizasyonu
İlaçlar: Prostoglandin türevleri, epinefrin, nikotinik asit, dosetaksel, paklitaksel, tamoksifen.
Kalıtımsal retina distrofileri: Retinitis pigmentoza, otozomal dominant kistoid makula ödemi.
Traksiyonel makülopatiler: VMT sendromu, ERM, miyopik traksiyon makulopatisi.
Göz içi tümörler: Koroidal kavernöz hemanjiom, retinal hemanjioblastom ve koroid melanomu.
Optik sinir başı anomalileri: Optik pit, kolobom (23).
Tanı Yöntemleri
- Oftalmoskopi: Yarıklı lamba biyomikroskobi muayenesi ile makulanın kalınlaştığı, foveal çöküntünün kaybolduğu ve kistik boşlukların varlığı izlenebilir (Resim 11).
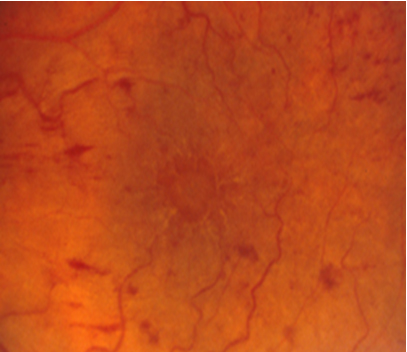
Resim 11: Kistoid makula ödemi olan hastanın renkli fundus görüntüsü. Makuladaki kistik boşluklar balpeteği görünümünde izlenmektedir.
- Floresein Anjiografi (FA): FA, KMÖ tanısında yaygın olarak kullanılan bir tanı yöntemidir. Erken dönemde sızıntıya bağlı hiperfloresan noktalar, geç dönemde ise kistik boşluklarda floresein birikimine bağlı KMÖ için karakteristik olan taç yaprağı görünümü izlenir (Resim 12). FA makula ödeminin tedavisini planlamak açısından da yardımcı bir testtir (24,25).
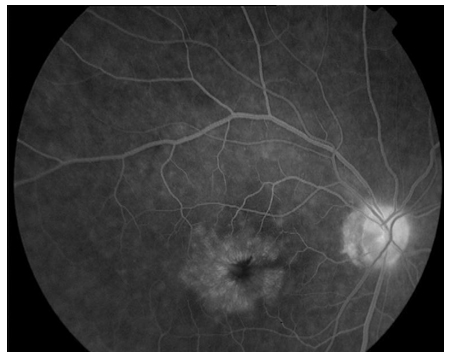
Resim 12: Irvine Gass Sendromlu bir olguda, flöresein anjiografide, KMÖ ye bağlı geç dönemde tipik taç yaprağı görünümündeki anjiografik sızıntı ile birlikte optik sinir başında boyanma dikkat çekmektedir.
- Optik Koherens Tomografi (OKT): OKT’nin günümüzde tanı açısından biyomikroskobi ve FA’ye üstünlüğü gösterilmiştir (26). OKT’de retinada kalınlaşma, siyah-reflektif olmayan kistik boşluklar ve bu kistik alanları çevreleyen orta-düşük reflektivitede septalar izlenir (27) (Resim 13)
Resim 13: Diyabetik retinopatiye bağlı bir KMÖ. OKT’de hiporeflektif kistoid boşluklar, retinada kalınlık artışı ve foveal çöküntünün kaybı izlenmekte.
Tedavi
Non-steroid antiinflamatuar damlalar (NSAİD), karbonik anhidraz inhibitörleri (KAİ), steroidler (topikal, perioküler enjeksiyon, intravitreal enjeksiyon, sistemik), immünmodülatör tedavi, hiperbarik oksijen tedavisi medikal tedavi yöntemleridir. Topikal NSAİD ve steroid damlalar ve katarakt cerrahisi sonrası gelişen KMÖ’nün tedavisinde kombine kullanıldıklarında semptomların daha hızlı gerilediği gösterilmiştir, tedavi doz azaltılarak kesilmelidir. Asetazolamid ve diğer KAİ’nin topikal ve sistemik kullanımı, Irvine Gass Sendromu ve RP kaynaklı KMÖ tedavisinde etkili olabilmektedir. Sistemik steroid ve immünmodülatör tedavi (Siklosporin, metotreksat, azotioprin) üveite ikincil gelişen KMÖ’de kullanılır (21).
İntravitreal anti-vasküler endotelyal growth faktör ajanlar (antiVEGF) özellikle diyabetik retinopati ve retina ven tıkanıklığı gibi retinal vasküler hastalıklara ikincil KMÖ’nün tedavisinde etkili bulunmuştur. Günümüzde KMÖ tedavisinde kullanılan başlıca anti-VEGF ajanlar ranibizumab, bevacizumab ve aflibercepttir. Ayrıca üveit ve radyasyon retinopatisine bağlı gelişen KMÖ tedavisinde de anti-VEGF ajanlar etkilidir (28).
Lazer ve vitrektomi, KMÖ tedavisinde kullanılan cerrahi yöntemlerdir. Diyabetik KMÖ ve retina ven dal tıkanıklığına ikincil KMÖ’de fokal ve grid lazer fotokoagülasyonu kullanılabilir. Son yıllarda mikropulse laser de etkin ve güvenli bir seçenek olarak gündeme gelmiştir. Vitreomaküler traksiyonun etken olduğu durumlarda vitrektomi düşünülmelidir.
SANTRAL SERÖZ KORYORETİNOPATİ
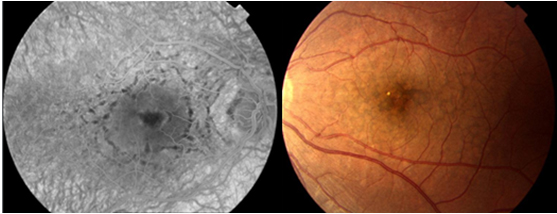
Santral seröz koriyoretinopati (SSKR), retina pigment epitelinin (RPE) bir veya daha fazla fokal lezyonu birlikteliğinde nörosensoriyel retinanın iyi sınırlı seröz dekolmanı ile karakterize idiopatik bir hastalıktır (Resim 14).
Resim 14: Sol gözde SSKR’si olan hastanın nörosensöriyel seröz dekolman alanı izleniyor.
SSKR, sıklıkla 25-55 yaşları arasındaki genç erkeklerde tek taraflı olarak karşımıza çıkmaktadır. Risk faktörleri arasında; psikolojik veya fiziksel stres, A tipi kişilik, glukokortikoid kullanımı, sistemik hastalıklar (SLE, Cushing), gebelik, aile öyküsü varlığı ve beyaz ırk bulunmaktadır (29).
Semptomlar; santral skotom, metamorfopsi, diskromatopsi, mikropsi, kontrast duyarlılığında azalma ve bulanık görmedir. Dekole olan duyusal retina öne doğru yer değiştirdiği için hipermetropi meydana gelir. En iyi düzeltilmiş görme keskinliği 20/20 ile 20/200 arasında değişir, akut vakaların %75’inde en iyi düzeltilmiş görme keskinliği 20/30 ve daha iyidir (30).
Fundus muayenesinde, akut hastalıkta tipik olarak arka kutupta sınırları belirgin oval-yuvarlak şekilli nörosensöryel retina dekolmanı izlenir. Beraberinde seröz pigment epitel dekolmanı (PED) da bulunabilir. Normal foveal reflenin kaybı tanı koymada yardımcıdır. Kronik tekrarlayan olgularda, RPE değişiklikleri ve atrofisi görülebilir. SSKR seyrek olarak büllöz nörosensöryel dekolman ile karşımıza gelebilir, bu klinik tablo genellikle inferior yerleşimlidir ve yerçekimi etkisi ile subretinal sıvının makuladan aşağıya doğru drene olduğu izlenir (31). Bu tablo gerilediğinde geriye yaygın, inferior perifere uzanan RPE atrofileri görülür (Resim 15)
Resim 15: Yaygın RPE atrofileri ve inferior perifere uzanan yaygın pencere defektleri bulunan atipik SSKR olgusunun FA görüntüsü.
Tanı Yöntemleri
1.Floresein Anjiografi (FA): Akut SSKR’de, RPE seviyesinde fokal floresein sızıntısı tipik FA bulgusudur. Mürekkep lekesi görünümü, SSKR vakalarında en sık görülen sızıntı paternidir (Resim 16 ). Noktasal sızıntı FFA’nın erken fazında belirir ve yavaş yavaş simetrik biçimde tüm yönlere yayılır. Tüten baca görünümü ise en tipik FA paternidir, yaklaşık %10 oranında görülür, erken dönemde izlenen hiperfloresan nokta baca dumanı şeklinde şeklinde dekolman alanının en üst noktasına kadar yükseldikten sonra nazal ve temporal alanlara yayılarak mantar şeklini alır (Resim 17) (29).
Resim 16: FA’de Mürekkep lekesi paterninde erken ve geç dönem hiperfloresans izlenmekte.
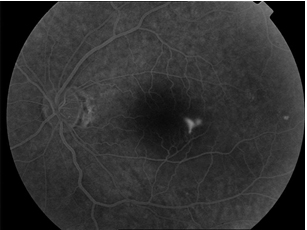
Resim 17: SSKR’si olan hastanın FA’de tüten baca paterninde hiperfloresans izleniyor.
2.İndosiyanin Yeşil Anjiografi (İYA): SSKR’de koroid damar geçirgenliği artmıştır, buna bağlı olarak İYA’de orta evrede hiperfloresans izlenir.
- Optik Koherens Tomografi (OKT): Akut SSKR olgularında OKT’de nörosensöryel retina dekolmanı ve eşlik ediyorsa PED izlenir (Resim 17) . Kronik olgularda SSKR olgularında fırçamsı kenar oluşumu fotoreseptör dış segment lokalizasyonu ile uyumlu olarak izlenir. Seröz dekolmanın uzun süre devam ettiği olgularda uzayan fotoreseptör dış segmentlerinin zamanla subretinal boşluğa ve RPE üzerine döküldüğü ve OKT’de subretinal boşlukta hiperintens granüler birikimler olarak izlenebilir (resim 18). EDI-OKT’nin kullanıma girmesi ile SSKR’de subfoveal koroid kalınlığında artma olduğu gösterilmiştir (Resim 19)(31).
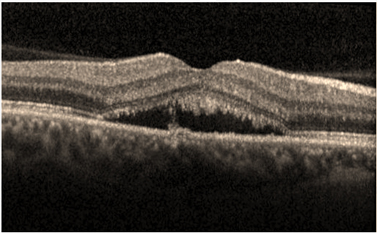
Resim 18: SSKR olgusunda OKT’de seröz retina dekolmanı ve uzun süreli olduğunu gösteren bir bulgu olarak fırçamsı kenar oluşumu.

Resim 19: SSKR hastasında EDI-OKT de koroidin oldukça kalın olduğu görülmektedir. Koroid kalınlığı bu olguda 462 micron olarak ölçülmüştür.
Tedavi
SSKR, benign seyirli bir hastalıktır, genellikle 1-4 ay içerisinde kendiliğinden iyileşme görülür ve görme keskinliği normal seviyelere yükselir. Ancak, hastaların %50’sinde nükslerle seyreder. Akut SSKR’de ilk yaklaşım, hastayı gözlemek ve risk faktörleri varsa ortadan kaldırmaktır (29).
Fokal Argon lazer fotokoagulasyonu, FA’de saptanan sızıntı alanlarına uygulanır (Resim 20). Subretinal sıvının rezolüsyonunu hızlandırsa da sonuç görme keskinliği ve nüks sıklığı üzerine bir etkisi olmadığı gösterilmiştir (29). Bu tedavi daha ziyade extrafoveal az sayıda sızıntı odağı olan hastalarda tercih edilebilecek bir yöntemdir.
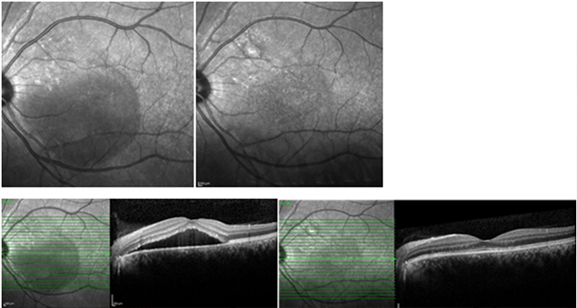
Resim 20: SSKR nedeniyle seröz dekolmanında 5 ay içinde gerileme olmayan bir doktor hastamızda tek odaktan olan sızıntı fokal laser ile tedavi edildikten sonra hızla dekolmanın çekildiği ve 1 ay içinde tamamen yokolduğu görülmekte. Üstte infrared otofloresans görüntüde solda lazer öncesi, sağda ise sonrası izlenmekte. Bu görüntüde üst temporal arkuat içinde fovea üst nazalinde lazer skarı (üst sağ) görülmektedir. Altta solda tedavi öncesi, sağda ise sonrasındaki OKT görüntüsü izlenmektedir.
Fotodinamik tedavi (FDT), yeni bir tedavi seçeneğidir kronik olgularda ve fotokoagulasyondan uzak durulması gereken jukstafoveal ya da subfoveal lokalizasyonlu RPE lezyonlarında daha sıklıkla kullanılmaktadır (29, 30). İndosiyanin yeşil (ICG) anjiografide tespit edilen koroidal hiperpermiabilite alanları hedef alınarak uygulandığında FA rehberli uygulamalara göre daha başarılı sonuçlar elde edilmektedir. Genellikle standart uygulamadan ziyade yarı dozda verteporfrin ile veya yarı güçte diode laser ile uygulamalar tercih edilmektedir. Tedaviye cevap genellikle oldukça iyidir ve başarı oranları yüksektir. Bu haliyle kronik SSKR de ilk tercih edilebilecek tedavi seçeneğidir denilebilir.
İntravitreal anti- vasküler endotelyal büyüme faktörü (anti-VEGF) enjeksiyonu da tek başına ya da FDT ile birlikte SSKR tedavisinde yararlı olabileceğini gösteren yayınlar olmakla birlikte bunlar çok küçük vaka serileridir (31).
OPTİK PİT MAKÜLOPATİ
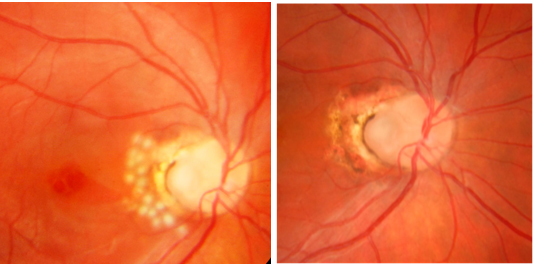
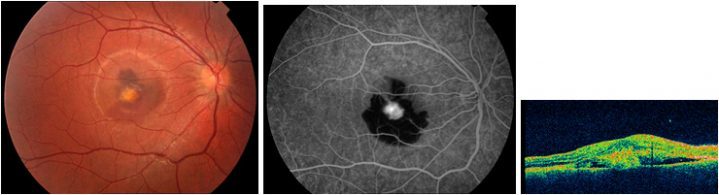
Optik pit, Wiethe tarafından 1882 yılında tanımlanmış olup, nadir görülen konjenital bir anomalidir. Optik pitin optik fissurün tamamlanmamış kapanması nedeniyle olduğu düşünülmektedir. Sıklıkla optik diskin inferotemporal bölgesinde yerleşimlidir. Sıklıkla asemptomatik olup, görmede azalma yapmamaktadır. 11.000 de bir görülen bu anomali %15 oranında bilateral olabilmektedir. Sıklıkla asemptomatik olan bu anomali, maküler ödem, dış retinal katlarda skizis, dış lameller hol, seröz makula dekolmanı gibi makulopati kliniğine yol açarak görme keskinliğinde azalma, santral skotom gibi semptomlara yol açmaktadır (32) (Resim 21).
Optik pitin embriyonal gelişim sırasında optik fissürün inkomplet kapanması sonucu geliştiği düşünülmektedir. Pit alanında retina sinir lifi kaybı da mevcuttur. Optik pit makulopatisinin (OPM) patofizyolojisi henüz tam anlaşılamamakla birlikte pek çok hipotez öne sürülmüştür. Bunlardan biri Brown ve ark. tarafından öne sürülen retina altı sıvının vitröz kaviteden kaynaklandığı varsayımıdır. Diğer bir hipotez retina altı sıvının subaraknoid boşluk aracılığıyla serebrospinal sıvıdan kaynaklandığını öne sürmüştür. OKT ile yapılan çalışmalarda subaraknoid boşluk ile subretinal boşluk arasında direkt bir ilişki olduğu görülmüştür. Retina altı sıvının sızdıran bazı damarlardan kaynaklandığını veya orbital boşluktan kaynaklandığını ileri süren hipotezler de mevcuttur (33).
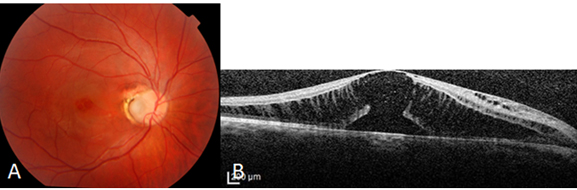
Resim 21: Optik pite bağlı seröz makula dekolmanı ve tipik OCT bulguları görülmektedir. Özellikle dış retinal retinoskizis ve dış lamellar hol çok demonstratif olarak görülmektedir.
Tedavi:
Optik pit veya koloboma bağlı gelişen makulopatilerde genellikle benzer tedavi stratejileri uygulanmaktadır. Kabul görmüş tek bir yöntem bulunmamaktadır ve uygulanmakta olan tüm tedaviler tartışmalıdır. Anlatılacak tedavi yöntemleri hem optik pitte hem de optik kolobomda uygulanmakta olan yöntemlerdir.
İlk olarak konservatif gözlem yaklaşımı pek çok merkez tarafından önerilmektedir (34). Gelişen makulopatilerin %25’inin kendiliğinden düzelebildiği istatistiği göz önüne alınırsa ilk başvuruda 3 ay tedavisiz gözlem uygun olmaktadır. Fakat kendiliğinden düzelen seröz makula dekolmanı olgularında son görme keskinliği daha düşük olmaktadır (35).
1969 yılında Gass, optik disk temporal kenarına Xenon lazer fotokoagülasyon uygulayarak optik disk temporal kenarında koryoretinal adezyon sağlayıp pit alanından retina altına sıvı geçişini engelleme fikrini ortaya atmış, fakat uygulamalarında yüz güldürücü sonuçlar alamamıştır (36). Uzun yıllar optik pit/koloboma bağlı makulopatilerde 3 aylık izlemden sonra düzelmeyen hastalarda ilk tedavi olarak Argon lazer fotokoagülasyon uygulanmıştır (Resim 22). Argon lazer fotokoagülasyon anatomik başarı sağlanması için uygun olabilecek bir metod olmasına rağmen bu başarı için gereken sürenin kestirilememesi ve fonksiyonel başarısı düşük olması nedeniyle en iyi tedavi seçeneği olmaktan uzaktır.
Resim 22: Optik pitli bir olgumuzda optik disk temporaline uygulanmış 2 sıra lazer fotokoagülasyonun erken ve geç dönem görünümü.
Argon lazer fotokoagülasyon ile birlikte veya tek başına intravitreal gaz verilmesi de uygulanan tedavi metodlarından birisidir. Lincoff ve ark. intravitreal gaz tamponad ile makula altı sıvının yer değiştirmesinin sağlanabileceği ve görme keskinliğinde artış elde edilebileceğini öne sürmüştür (37). Akiyama ve ark. 8 hastalık bir seride ortalama 1.8 kez %100 sulfur hexafluoride 6 (SF6) gaz enjeksiyonu ile %50 anatomik ve fonksiyonel başarı sağlamışlardır (38). Bu çalışmada takip süresi kısa olduğu için bu başarının kalıcı olup olmadığı bilinmemektedir. Gaz enjeksiyonuna rağmen retina iç tabakalara sıvı akışının engellenmediği göz önüne alınırsa etkinin kalıcı olmayabileceği düşünülebilir.
Son yıllarda optik pite bağlı makulopati tedavisinde en sık uygulanan yöntem pars plana vitrektomidir (PPV). PPV’de başarının vitreus alınarak optik pit üzerindeki traksiyonel kuvvetlerin ortadan kaldırılması ile makula altına pasif sıvı geçişini azaltması yoluyla olduğu düşünülmektedir. PPV’deki esas amaç posterior hyaloidin kaldırılmasıdır. Bazı cerrahlar bunu kesinleştirmek için iç limitan membran soyma (ILMP) işlemini de gerçekleştirirler. Fakat bazı yazarlar bunun gerekli olmadığını anatomik ve fonksiyonel başarı için arka hyaloidin tam olarak kaldırılmasının yeterli olduğunu savunurlar (39). Avcı ve ark. ortalama 16 ay takip ettikleri 13 hastalık bir seride 12 hastada ILMP yapmadan arka hyaloidin tamamen kaldırılmasıyla yapılan PPV ile anatomik ve fonksiyonel başarı sağlamışlardır (40). Post-op dönemde subretinal sıvının çekilmesi için sabırlı davranmak gereklidir, zira sıvının çekilmesi aylar alabilir.
PPV ile birlikte gaz tamponadın da sonuca etkisi olup olmadığı tam olarak bilinmemekte ve gaz tamponad kullanılmadan yapılan cerrahilerde de başarılı sonuçlar alınmaktadır.
Optik pitten direkt sıvı geçişinin engellenmesi için de çeşitli çalışmalar yapılmıştır. Rosenthal ve ark. optik pit üzerine otolog platelet enjekte ederek koagülasyon faktörlerinin çalışmasını indüklemeyi, bu şekilde optik pitten serebrospinal sıvının geçiş yollarının kapanmasını hedeflemişlerdir. Yayınladıkları vaka raporunda anatomik ve fonksiyonel başarı sağlasalar da henüz uzun dönem sonuçları bilinmemektedir (41).
Travassos ve ark. ise optik pit ağzına alt nazal kadrandan alınmış yaklaşık 0.5 mm3 boyutlarında skleral dokuyu yerleştirerek optik piti kapatmaya çalışmışlardır. PPV sonunda makula altı sıvı drene edilip skleral doku ile optik pit ağzı kapatılmış ve gaz tamponad verilmiştir. Gaz tamponad olarak %14 C3F8 kullanılmıştır. 3 hasta içeren vaka serisinde bütün hastalarda anatomik başarı elde etmişlerdir (42).
Optik pit alanını kapatmak için öne sürülen bir başka yöntem ise ILM’nin soyulup yerinden ayırmadan OSB etrafında flap haline getirilerek ve OSB üzerine döndürülerek pit üzerine kapatılmasıdır. Bu teknikte standart PPV sonrasında OSB etrafında oluşturulan ILM flapleri ters çevrilerek pit üzerine kapatılmaktadır (inverted flap tekniği). Bu yolla subaraknoid boşluk ile subretinal boşluk arasındaki bağlantının engellenmesi planlanmıştır. Mohammed ve ark. yayınladığı bir olgu sunumunda bu teknik ile anatomik ve fonksiyonel başarı sağlamışlardır (43).
Diğer bir tıkayıcı madde ise otolog fibrin yapıştırıcısıdır. Otolog olması nedeniyle reaksiyon riskinin olmaması, kuvvetli bir adezif biyomolekül olması nedeniyle subaraknoid boşluktan subretinal alana sıvı geçişini engelleme potansiyeli sayesinde başarı şansını yüksek oranda arttırabilecek bir biyomoleküldür. Otolog fibrin yapıştırıcısı kan bankalarında özel cihazlar ile hazırlanabilmektedir. Hastadan sitrat eklenmiş tüpe alınan yaklaşık 120 ml kan ile 5 ml otolog fibrin yapıştırıcısı hazırlanabilmektedir. Fibrin yapıştırıcısı dış ortamda beklememesi gerektiği için operasyon günü alınan kandan hazırlanıp hemen arkasından operasyonda kullanılmaktadır. Otolog fibrin yapıştırcısı ile birlikte koaktivatör (pH10 buffer solüsyonu) kullanılarak tepkime hızlandırılmaktadır (44).
Bu yöntem kliniğimizde kullanılarak dünyada ilk kez tanımlanmış ve literatüre kazandırılmış bir teknikdir (45). Bu tekniği vitrektomiye rağmen nüks eden persistan makuler dekolmanlı olgularda kullanmaktayız. Bu olgularda sıvı hava değişimi sonrası optik sinir başına (OSB) önce bir damla fibrin, ardından bir damla koaktivatör damlatılmakta ve saniyeler içinde fibrin oluşumu görülmektedir. Genellikle OSB dışına taşan fibrin fazlası hava sıvı değişimi sonrası soyulmakta ve sadece OSB üzerindeki kısım bırakılmaktadır. Sonrasında sıvı hava değişimi ile cerrahi sonlandırılmaktadır. Postoperatif OSB OCT kesitlerinde de net olarak fibrin materyali görülebilmektedir (Resim 23).
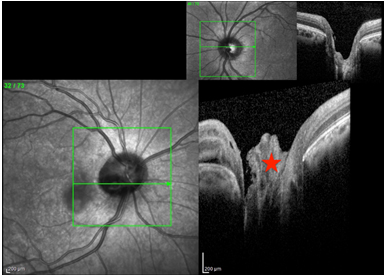
Resim 23: Post-op 8. ayda optik sinir başından alınan OKT görüntüsünde kolobom alanını dolduran otolog fibrin yapıştırıcısı görülmektedir (kırmızı yıldız). Sağ üst köşede ise diğer gözde aynı alan boşluk olarak görülmektedir.
İDYOPATİK KOROİD NEOVASKÜLARİZASYONU
İdyopatik koroidal neovaskülarizasyon (İKNV), 50 yaş altını etkileyen nadir görülen bir klinik tablodur. İKNV’de; koroidal neovaskülarizasyona neden olan yaşa bağlı dejenerasyon, miyopi, histoplazmozis, anjioid streaks, inflamatuar, travmatik ya da herediter durumlar gibi altta yatan herhangi bir maküla ya da retina hastalığı bulunmamaktadır.
İKNV membranlar genellikle kendi kendini sınırlar, hastalığın doğal seyri ve görme prognozu yaşa bağlı makula dejenerasyonu ya da patolojik myopi sonucu gelişen KNV’den daha iyidir (46,47). Ancak tedavi edilmediği durumda ciddi görme kaybı gelişir. Bu nedenle çeşitli tedavi yöntemleri denenmiş olmakla birlikte bilinen en etkin tedavi yöntemi intravitreal anti-VEGF tedavidir.
İKNV olgularında intravitreal bevacizumab enjeksiyonu ile görme keskinliğinde artış ve merkezi makula kalınlığında incelme olduğu bildirilmiştir (48,49). Görme prognozu yaşa bağlı makula dejenerasyonuna göre daha iyi olup genellikle tedavi sayısı da daha azdır. Bu durum daha genç hastada ortaya çıkması nedeniyle RPE’nin daha sağlıklı olmasıyla açıklanabilir.
PATOLOJİK MİYOPİ
Fizyolojik miyopi dünya çapında sık rastlanan bir göz bozukluğudur. Patolojik miyopi, göz küresinin 26 mm’den uzun olması ve refraksiyon değerinin -6 Dioptriden (D) büyük olması ile fizyolojik myopiden ayrılır. Daha önceleri malign miyopi, dejeneratif miyopi gibi isimlerle anılan patolojik miyopide; miyopiye bağlı sklera, koroid ve retina pigment epitelinde oluşan dejeneratif bozukluklar sonucunda gözde kalıcı hasar oluşabilmektedir (50).
Günümüzde patolojik miyopinin kalıtsal yönü ve sistemik hastalıklarla birlikteliği bilinmektedir. Down sendromu, Marfan sendromu, Noonan sendromu, prematürite, Ehler-Danlos sendromu, Pierre-Robin sendromu ve Stickler sendromu gibi durumlarda bu sorunun görülme sıklığının arttığı gösterilmiştir (50).
Patolojik miyopide göz küresi ilerleyici bir şekilde ön-arka doğrultuda uzar ve göz yapılarının mekanik olarak gerilmesine ikincil değişiklikler görülür ve bu hastalar az görme şikayeti ile başvururlar.
Patolojik miyopların fundus muayenesinde;
- Vitreus likefaksiyonu,
- Arka vitreus dekolmanı,
- Koroidal veya skleral kresentlere yol açan peripapiller atrofiler, fokal veya diffüz koryoretinal atrofi alanları (Resim 24),
- Periferik retinada lattice dejenerasyonları,
- Miyopik konüs ve tilted disk görünümü; Miyopik dejenerasyonun oluşturduğu koryoretinal gerilme ve atrofi; optik sinir etrafında, alttaki skleranın görülebildiği, temporal yerleşimli bir kresentin oluşumuna yol açar, peripapiller atrofi olarak da bilinen bu bulgu patolojik miyopinin kardinal bulgularından biridir,
- Retina pigment epitel (RPE) atrofileri,
- Makuler alanda “Fuch’s noktası”; makülada oluşan subretinal hemorajinin çekilmesi ile oluşan yuvarlak, pigmente lezyon (Resim 25),
- Lacquer çatlaklarına yol açan Bruch membranı ve koryokapiller kırıkları,
- Arka stafilom; skleradaki zayıflığa bağlı arka kutupta görülen ektazidir ve armut gibi globun arka kısmında çıkıntı yapar (Resim 26) (51).

Resim 24: Patolojik miyopisi olan hastanın sağ gözünde fokal koryoretinal atrofi alanları ve peripapiller atrofi izlenmekte.
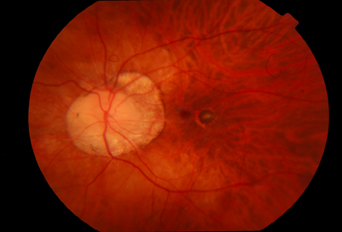
Resim 25: Patolojik miyopisi olan hastanın sol gözünde peripapiller atrofi (miyopik konüs) ve makulada Fuch’s noktası izlenmekte.
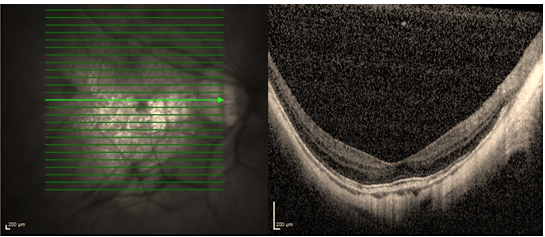
Resim 26: Posterior stafilomu olan hastanın OKT görüntüsü, maküler alandaki ektazi dikkat çekici düzeydedir.
Komplikasyonlar
Patolojik miyopide yukarıda sayılan fundus bulguları eşliğinde çeşitli komplikasyonların sıklığı da artmıştır.
Periferik retina yırtığı ve retina dekolmanı: Retinal yırtıklar genellikle arka vitreus dekolmanını takiben gelişmektedir. Kuvvetli vitreoretinal yapışıklıklar, zamanla tutunmakta oldukları yüzeyden ayrılarak lattice dejenerasyonunun arka kenarında ve 2 yanında yırtıkların oluşmasına yol açabilir. Işık çakması, sinek uçuşması ve perde inmesi gibi semptomları olan olguların detaylı fundus muayenelerinin hızlıca yapılması gerekmektedir. Sadece retinal yırtık veya delik gelişmiş olan olgulara laser fotokoagülasyon yapılması yeterli olurken, klinik olarak retina dekolmanı gelişmiş olanlarda cerrahi yöntemlerin tercih edilmesi gerekmektedir (52) (Resim 27).
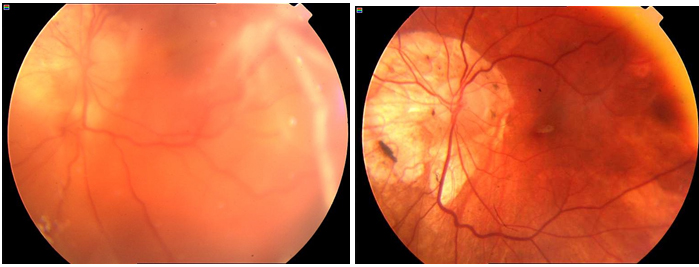
Resim 27: Patolojik miyopiye bağlı yırtıklı retina dekolmanı olan bir vakamızın preop (solda) ve vitrektomi sonrası postop (sağda) fundus görünümleri.
Miyopik foveoskizis ve makula deliği: Miyopik maküler retinoskizis ya da miyopik traksiyon makulopatisi olarak da isimlendirilir. Yüksek miyopisi olan posterior stafilomlu hastaların %9’unda bulunur ve %50’sinde 2 yıl içinde tam kat makula deliği veya makuler retina dekolmanı gibi daha ciddi komplikasyonlara ilerler. OKT ile maküler alanda retina tabakalarının birbirinden ayrıldığı izlenir (52). Miyopik makula deliğinin tedavisi idiyopatik makula deliğine göre nisbeten daha zordur ve prognozu daha kötüdür. Standart tedavi ile sonuç alınamayan vakalarda makulaya skleral çökertme materyali (buckle) uygulanabilir (Resim 28).
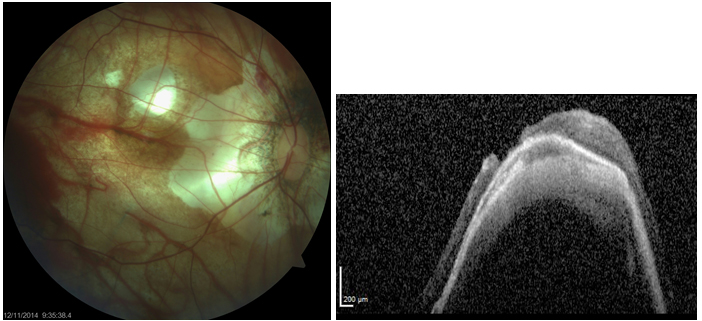
Resim 28: Patolojik miyopiye sekonder makula deliği ve makuler dekolman olan bir olgumuzda vitrektomi-ILMP’ye rağmen sonuç alınamadığından maküler buckle cerrahisi uygulanarak retinanın yatışması sağlanmıştır. Yukarda postop fundus görünümü ve OKT si izlenmekte.
Miyopik koroid neovaskülarizasyonu (KNV): KNV patolojik miyopisi olan hastalarda görme kaybına neden olan en sık komplikasyonlardan biridir. Yüksek miyopisi olan hastaların %10’unda KNV geliştiği bildirilmiştir. Kliniği, gri rengi, keskin sınırları, OKT’de pek fazla intraretinal-subretinal sıvı olmaması, FA da belirgin geç sızıntı yapmaması ile YBMD kliniğinden biraz farklıdır. Miyopik KNV prognozu genellikle orta düzeydedir. YBMD’ye göre daha iyidir ve daha az sayıda anti-VEGF enjeksiyonuna ihtiyaç duyulur. Bir gözünde miyopik KNV’si olan olguların sağlam gözünde de KNV gelişme insidansının arttığı bilinmektedir (51,52).
TRAVMATİK MAKULOPATİ
Kommosyo retina, Berlin ödemi olarak da adlandırılır, künt göz travmalarını takiben gelişen sensöryel retinanın ödemidir, makulayı tutarak akut travmatik makulopati tablosuna neden olabilir. Makuler alanda retinanın beyazlaşması ile karakterizedir, “makulada Japon bayrağı” görünümü izlenebilir. Görme kaybına neden olabilir. Travmanın ve hasarın ciddiyetine bağlı olarak, haftalar içinde kendiliğinden sekel bırakmadan gerileyebilir. Ciddi travmalarda makulada pigmenter dejenerasyon ve makula deliği oluşumuna ilerleyebilir (53).
Purtscher makulopatisi, Purtscher tarafından kafa travması sonrasında retinada meydana gelen, yama şeklindeki beyazlaşmalar ve kanamalar olarak tanımlanmıştır. Benzer görünüm göğüs ve kafa travması yanında; uzun kemik kırıkları, akut pankreatit, doğum eylemi, perioküler steroid kullanımı ve kollajen doku hastalıkları gibi pek çok hastalıkta da görülebilmektedir. Kafa travması haricindeki sebeplerle gelişen tablo bazı kaynaklar tarafından Purtscher benzeri sendrom olarak da adlandırılmaktadır. Purtscher retinopatisinin gelişiminde, küçük arteriollerde embolizasyona bağlı gelişen oklüzyonun ve sekonder enfarktın etkili olduğu bilinmektedir. Klinik olarak tablo kendisini akut ve ağrısız görme kaybı ile gösterir. Olguların çoğunluğunda hastalık bilateral olarak görülse de, nadiren tek taraflı olarak da teşhis edilebilir (54).
Travma sonrası makulada görülebilen diğer bir tablo koroid rüptürüdür. Koroid rüptürü koroid, Bruch membranı ve retina pigment epitelini içerir. Yeni olgularda subretinal kanama rüptürü maskeleyebilir (Resim 29). Makulayı içeren koroid rüptüründe görme prognozu kötüdür. Geç dönemde rüptür zemininde koroid neovaskülarizasyonu gelişebilir (53).
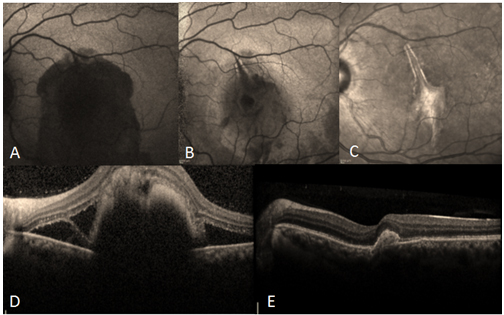
Resim 29: Travmaya bağlı koroid rüptürü olan hastanın sol göz fundus otofloresans (FOF) ve OKT görüntüleri, (A) hastanın başvuru anındaki FOF görüntüsü, makülada subretinal hemoraji, görme keskinliği 1 mps, (B) hastaya intravitreal 0,3 cc SF6 gazı ve doku plazminojen aktivatörü (tPA) enjeksiyonu + yüzüstü pozisyon verilmesini takiben 4.gün FOF görüntüsünde subretinal hemorajinin büyük oranda çekildiği izleniyor, görme keskinliği 0.2, (C) hastanın postoperatif 1.ay FOF görüntüsü, hemoraji çekilmiş, görme keskinliği tam. (D) hastanın preoperatif ve (E) postoperatif OKT görüntüleri.
HEREDİTER MAKULA DİSTROFİLERİ
Best Hastalığı ( Best Vitelliform Distrofi, Best Makula Distrofisi)
Best makula distrofisi (BMD), BEST1 geninde mutasyonlara bağlı gelişen otozomal dominant (OD) geçişli bir hastalıktır. Aile hikayesi sıklıkla pozitifdir. 1/10000 sıklıkta görülür. Primer olarak retina pigment epitelinin (RPE) etkilendiği bir maküler hastalıktır.
Rutin oftalmoskopik muayanede genellikle tipik vitelliform lezyonlara rastlanır. Kliniği erken evrelerde (vitelliform) oldukça tipiktir. Genellikle bilateraldir. Klasik vitelliform lezyon yumurta sarısı, bazen portakal renginde, yuvarlak, bir miktar eleve ve etrafı koyu sınırla çevrilidir (Resim 30). Makuladaki diskoid lezyon 0.5-3.0 disk çapında olabilir, retinal damarlar diskin kenarından etkilenmemiş bir şekilde geçerler. Makuladaki lezyon sahanda yumurta görüntüsünü andırmaktadır (55).
BMD’nin evreleri şöyle sıralanabilir:
- Normal fovea (patolojik elektrookülogram-EOG)
- Previtelliform evre
- Vitelliform evre (Resim 30)
- Psödohipopiyon evresi (Resim 31)
- Çırpılmış yumurta evresi (vitellirüptif evre) (Resim 32)
- Korioretinal atrofi evresi
Previtelliform evrede fundus normal görünümdedir, EOG bozulmuştur, görme korunmuştur. Vitelliform evrede BMD’nin tipik bulgusu olan makulada yumurta sarısı şeklindeki yuvarlak lezyon görülür ve görme hala iyi düzeydedir. Çırpılmış yumurta evresinde lezyondaki sarı renkli materyal parçalanıp çırpılmış yumurta görünümü oluşturur. Bu noktada genellikle hastanın görmesinde azalma da başlar. Disk içeriği parçalanabilir ve sıvı seviyesi veren bir kist oluşabilir. Hipopiyona benzerliği nedeniyle bu evreye psödohipopiyon evresi denir. Son evrede makulada korioretinal atrofi alanları bazen de RPE proliferasyonuna bağlı pigmente alanlar görülür (55).
Erişkinlerde görülen vitelliform lezyonlar patern distrofiler başlığı altında anlatılmıştır.
Elektroretinogarm (ERG) ve karanlık adaptasyonu normaldir. EOG bu hastalık için en spesifik tanı testidir ve subnormal olmasıyla tanı koydurucudur.
FFA’da vitelliform evrede, koroid floresansının blokajına bağlı hipofloresans izlenir. Sarı pigment aşağıya çöktükçe üstte hiperfloresans görülür. Atrofik evrede erken hiperfloresans izlenir (Resim 30, 31, 32).
Fundus otofloresans (FOF) incelemesinde vitelliform lezyon tipik olarak hiperotofloresandır, psödohipopiyon evresinde hipopiyon görüntüsüne denk gelen alanlarda hiperotofloresans izlenir (Resim 33). Atrofik evrede hipootofloresans görülür (56).
OKT’de vitelliform evrede RPE altında homojen vitelliform materyal birikimi izlenir (Resim 33). Atrofik evrede yaygın RPE değişiklikleri ve fotoreseptör hücrelerde dejenerasyon görülür.
Previtelliform ve vitelliform evrede görme normal ya da hafif azalmıştır, ileri evrelerde görme 0.2-0.4 seviyelerine düşer ancak görme keskinliği genellikle dramatik klinik görünüme kıyasla iyidir. Genellikle iki göz arasında asimetrik tutulum söz konusudur ve bir gözde okumaya yetecek düzeyde bir görme ileri yaşlara kadar korunmaya meyillidir.
Hastalığın yıkıcı dönemlerinde arka kutupta görme keskinliğini azaltan koroid neovasküler membran gelişebilir veya KNV ile ilişkili olmayan subretinal kanama gibi eksüdatif değişiklikler ortaya çıkabilir.
Ancak koroid neovasküler membran gelişmesi çok daha nadir bir komplikasyondur. Gelişen neovasküler membranın tanısında FA ile lezyona ait sızıntı görülür. OKT’de neovaskülarizasyona bağlı artmış yansıtıcılıkla birlikte subretinal/intraretinal sıvı mevcuttur (Resim 34) . KNV’nin tedavisinde FDT, lazer fotokoagülasyon ve intravitreal anti-VEGF enjeksiyonu etkilidir (57).
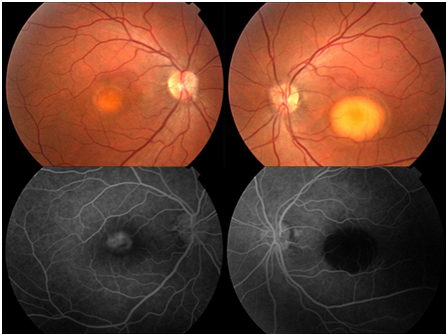
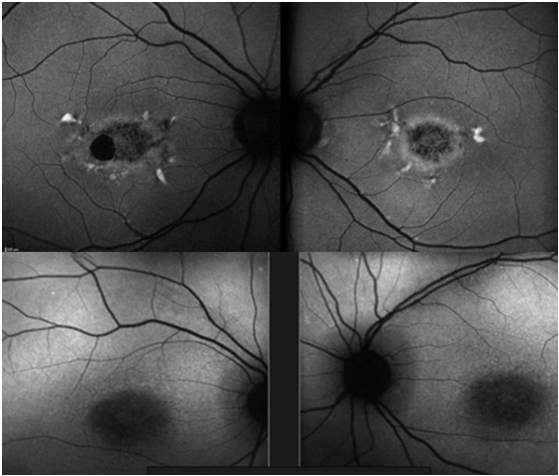
Resim 30: Best hastalığı, sağ gözde vitellirüptif evrede makuler lezyon, sol gözde vitelliform evrede tipik yumurta sarısı şeklinde yuvarlak makuler lezyon ve FA’de sağ gözde hiperflöresans, sol gözde bloke flöresans nedeniyle hipoflöresans izlenmekte. EOG de Arden oranları 1.2 ve 1.1 olarak elde edilmiştir.
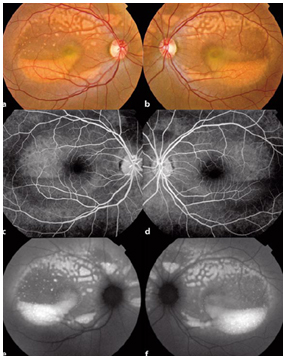
Resim 31: Best hastalığı, Psödohipopyon evresine ait RFF, FA ve FOF görüntüsü
Resim 32: Best hastalığı, vitellirüptüf evredeki makula lezyonu ve FA görüntüsü.
Resim 33: Best Hastalığı, (A) Sağ göz makuler lezyona ait OKT kesiti, santralde vitelliform materyal eriyip yerini subretinal boşluğa bırakmış, (B) lezyona ait daha inferior bir kesitte vitelliform materyal hiperreflektif olarak izleniyor (C) sol göz makuler lezyona ait OKT kesiti, (D) ve (E) sağ ve sol göz FOF testinde makuler lezyonlarda vitelliform madde birikimine bağlı hiperotofloresans izleniyor.
Resim 34: KNV’ye sekonder submaküler hemoraji ile komplike olmuş (1. sıra resimler) 12 yaşında bir Best olgusunda başlangıç görme 0.3 iken, PDT sonrası 2. Ayda (2. Sıra resimler) görmenin 0.9’a çıktığı, hemorajinin büyük oranda çekildiği ve OKTdeki subretinal sıvının yokolduğu izlenmekte. Bu durumun 23. Ayda da korunduğu 2. sıra aşağıdaki OKT görünümünden de görülmektedir.
Stargardt Makula Distrofisi ve Fundus Flavimakulatus
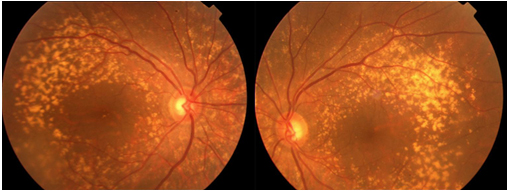
Stargardt’ın maküla distrofisi, Stargardt hastalığı, en sık görülen kalıtımsal makula hastalığıdır. Tüm retina distrofilerinin yaklaşık %7’sini oluşturmaktadır. Stargardt hastalığı genellikle otozomal resesif geçiş gösteren retina pigment epitelinde (RPE) lipofusin maddesi birikimi ile karakterize bir hastalıktır. Stargardt hastalığında ABCA-4 gen mutasyonu tespit edilmiştir. Stargardt hastalığı terimi sarı-beyaz sınırları belirsiz birikintiler (flekler-benekler) ile çevrili atrofik makula dejenerasyonu için kullanılır (Resim 35), flekler maküla ve arka kutupta yoğunluk gösterir. Bu fleklerin fundusta yaygın olarak yer aldığı klinik tablo ‘fundus flavimakulatus (FF)’ şeklinde isimlendirilmiştir (Resim 36). Daha sonra yapılan çalışmalar Stargardt hastalığı ve fundus flavimakulatusun iki farklı isimde tanımlanmalarına rağmen aynı hastalığın farklı tipleri olduğunu desteklemektedir (58).
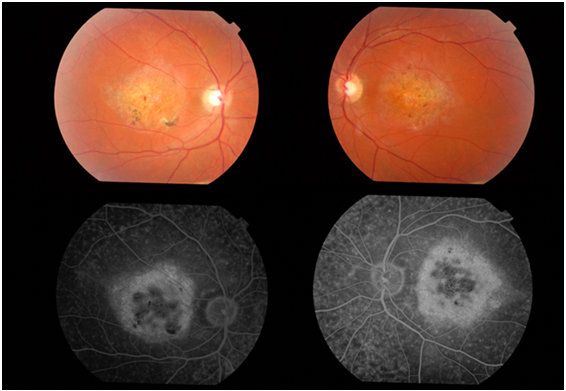
Resim 35: Stargardt hastalığı, bilateral makulada yaygın atrofi ve FA’de karanlık koroid görünümü.
Klinik Bulgular ve Prognoz
Hastalık belirtileri genellikle ilk dekadda ortaya çıkmaktadır. FF, Stargardt hastalığına göre daha ileri dönemde (3-4. dekadlarda) ortaya çıkar.
Stargardt hastalığında görme genellikle ilk dekadın sonu veya ikinci dekadın başında 1/10 veya daha düşük seviyelere düşer. FF’da ise görme nispeten daha iyidir. Görme çoğunlukla 40 yaşından sonraki dönemde okumayı zorlayacak sevilere düşer. Flekler foveayı tutmadığı sürece asemptomatik kalabilirler ancak bazı hastalarda Stargardt hastalığına benzer şekilde atrofik makulopati gelişebilir ve görme keskinliği azalır. Aynı ailenin farklı bireylerinde farklı fenotipler görülebilir (59).
Stargardt hastalığının erken evrelerinde oftalmoskopik bir bulgu olmayabilir, ancak zamanla santral atrofi alanlarını çevreleyen flekler izlenir. Makula bölgesindeki atrofiye uğramış RPE alanları birleşerek tipik ‘dövülmüş bakır manzarası’ görünümüne neden olmaktadır (Resim 35). FF hastalarında ise makula normal görünümde olup, sadece periferik yerleşimli flekler izlenebilir (Resim 36). Makuler atrofi ileri yaşlarda ve daha az oranda görülebilir.
FA’de en karakteristik bulgu, koroidin hipofloresan göründüğü ancak retina dolaşımının izlendiği, “karanlık koroid” ya da “sessiz koroid” görüntüsüdür. Bu bulgu hastaların 3/4’ünde bulunur ve tanı koymada yardımcıdır (Resim 35). Bu bulgunun nedeni RPE de biriken lipofusine bağlı bloke floresans görünümüdür. Nitekim zamanla bu hastalarda zamanla RPE hücrelerinde atrofi gerçekleşir (Resim 37).
ERG erken dönemde normaldir ancak ilerlemiş vakalarda yaygın RPE atrofisi geliştiğinde subnormal saptanabilir (60).
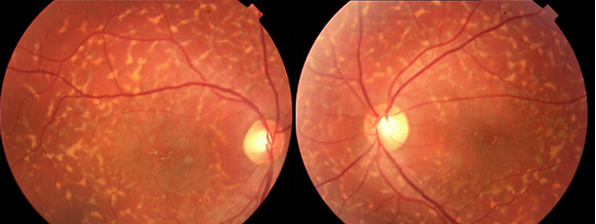
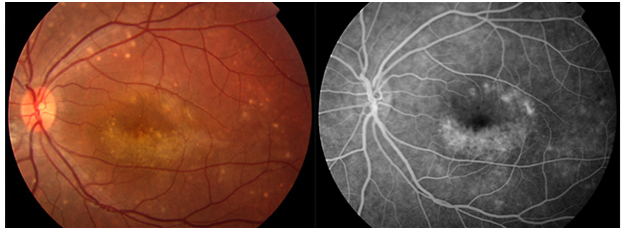
Resim 36: Fundus flavimakulatus’lu bir hastanın fundus görünümü. Bilateral tipik görünümde sarı flekler ve makülada RPE değişiklikleri izlenmekte. Bu dönemde hasta 27 yaşında ve görmesi bilateral 0.7 düzeyinde iken, hasta 44 yaşına geldiğinde santral atrofisinin ortaya çıktığı ve görmesinin 0.1/0.2 olduğu görüldü. Aynı hastanın kız kardeşinde ise 10 yaşından beri ciddi görme kaybı ile giden tipik Stargardt hastalığı görülmüştür.
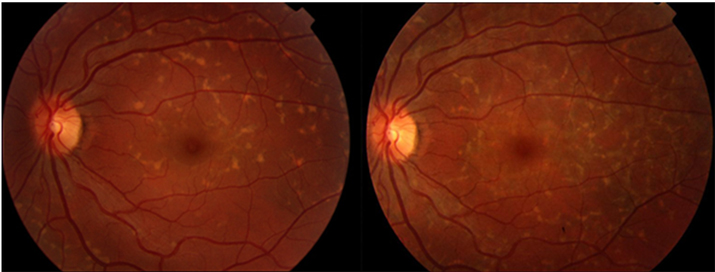
Resim 37: Fundus flavimakulatuslu bir hastanın 8 yıl arayla çekilmiş fundus fotoğrafında solda aktif olan fleklerin bir kısmının zaman içinde atrofiye dönüştüğü (sağ) izleniyor.
Fundus otofloresans (FOF) incelemesinde de tipik bulgular görülmektedir (Resim 38). Genel olarak RPE’de biriken lipofusine bağlı yaygın hiperOF ve peripapiller bölgede tipik peripapiller korunmuş bölge izlenir (Resim 38 alt). Aktif flekler hiperotofloresan (Resim 38 üst) inaktif flekler hipootofloresan ve atrofi alanları hipootofloresan olarak görülür. Stargardt hastalığında OKT de santralde dış retinal tabakalarda atrofi izlenmektedir (Resim 39).
Resim 38: Stargardt/FF da tipik FOF görüntüsü: Üstte flekler hiperOF, atrofi hipoOF olarak izlenmekte. Altta ise bir Stargardt hastasında FOF da tipik peripapiller korunmuş alan görülmekte.
Resim 39: Stargardt hastasında OKT de dış retinal tabakalarda ciddi atrofi görünümü.
Patern Distrofi
Retina pigment epitelinin (RPE) patern distrofisi, makuulada ve çevresinde gri veya sarı renkli pigment birikimi ile karakterize nadir görülen fundus değişiklikleridir. Bu pigment depozitleri farklı noktalar, çizgiler, dallanmalar şeklinde veya retiküler şekilde olabilir. Klasik patern distrofi (PD) biçimi, Deutman ve ark. tarafından 1970’de tanımlanmış olan kelebek şekilli distrofidir (KPD)(Resim 40). Daha sonra Gass, pigment dağılımının biçimine göre PD’yi 5 temel distrofi grubuna ayırmıştır: Erişkin başlangıçlı vitelliform makuler distrofi (AOVMD) (Resim 41), fundus pulverulentus (FP) (Resim 42), fundus flavimakulatusu taklit eden multifokal PD (FFPD), KPD ve retiküler distrofi (RD) (61).
Kelebek Şeklinde Patern Distrofi
Genellikle otozomal baskın kalıtım gösteren bir hastalıktır. Foveadan üç ayrı yönde uzanan sarı renkli pigmente lezyon izlenir. Foveolar ve foveal yüzeysel retina tabakalarının normal olduğu bilinmektedir. Optik disk, retinal damarlar ve koroid de normaldir (Resim 40).
ERG normaldir, EOG ise subnormal seyreder.
KPD, spesifik semptomunun olmayışı, ileri yaşlara kadar oldukça iyi santral görme düzeyinin korunması, normal renkli görme ve görme alanı ile karakterizedir. Ancak, hastalık yaşla birlikte ilerleyerek peripapiller bölgeye kadar uzanan atrofik, depigmente lezyonlara neden olabilir ve nispeten düşük olmakla birlikte ileri yaşlarda koroidal neovaskülarizasyon (KNV) riski bildirilmiştir. Her iki patoloji de santral görmeyi etkileyebilir ve dolayısıyla bu hastalığın en önemli prognostik faktörleridir (61).
Resim 40: Solda kelebek şekilli distrofinin (nokta, çizgi ve dallar biçiminde çeşitli şekiller oluşturan kelebeğe benzetilen pigment birikimi) anjiografik görünümü. Pigment birikimi anjiografide blokaj nedeniyle hipofloresan olarak izlenmektedir. Bloke floresans çizgilerinin etrafında sıklıkla var olan depigmentasyon nedeniyle hiperfloresan alanlar tipiktir.
Erişkin Başlangıçlı Vitelliform Makuler Distrofi
Makulada simetrik sarı depositler ile karakterizedir ve bu görüntüsüyle Best hastalığını andırır. Druzenle çevrili yumurta sarısı şeklinde maküler lezyonu olan hastaları ya da foveada beneklenme şeklinde sarı bir pigmentasyonu kapsayacak şekilde geniş bir tabloyu kapsamaktadır (Resim 41). Best hastalığından ayırıcı tanısı lezyonların daha küçük olması, EOG’nin normal ya da çok az azalmış olması, aile hikayesinin olmaması ve erişkin yaşta ortaya çıkması ile yapılır (62).
Erişkin hayatta görülen vitelliform lezyonlardan olan AOVMD yavaş ilerleyen bilateral görme kaybı ile seyreden, nadir bir makuuler hastalıktır. Lezyonlar 1/3-1/2 disk çapında, yuvarlak veya oval, hafif kabarık, gri-sarı renkli olup foveada çoğu zaman merkezi pigmentasyon ile birlikte bulunur. AOVMD tipik olarak 30-50`li yaşlarda görülür ve belirsiz genetik geçiş söz konusudur.
Ancak bazı otörler hastalığın otozomal dominant seyrettiğini, bazıları da sporadik olup ailesel bir geçiş paterni taşımadığını savunurlar. AOVMD`nin erken evrelerinde genellikle görsel açıdan asemptomatiktirler veya tek ya da çift taraflı hafif görsel bulanıklık veya metamorfopsiden şikayetçidirler. ETFVD ilerledikçe görme kaybı daha da şiddetlenir. Bunun nedeni vitelliform lezyonların bozulup yaşa bağlı makula dejenerasyonundakine (YBMD) benzer coğrafik atrofi oluşması veya daha az sıklıkla koroidal neovasküler membran (KNVM) gelişimidir (63).
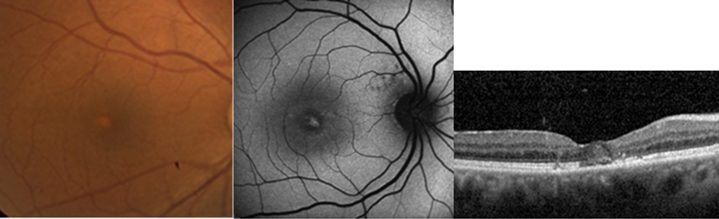
Resim 41: AOVMD: Sol başta makulada sarı renkli birikimin fundus görünümü, ortada başka bir olguda santralde hiperotofloresan birikim ve sağda bu hastanın OKT incelemesinde foveal vitelliform maddeye ait görünüm izlenmekte.
Resim 42: Fundus pulverulentus. Makulada RPE düzeyinde kaba, noktasal pigment birikintileri ve granüler fundus görünümü ile karakterizedir ve diğer paternlere eşlik edebilir.
Ailevi Dominant Druzen
Otozomal dominant geçişli bir hastalıktır ve çok geniş bir klinik spektrum gösterir. Başlangıçta asemptomatiktir ve görme keskinliği normaldir. Druzen rutin oftalmoskopik muayene sırasında saptanır. Görme keskinliğinde azalma, metamorfopsi sıklıkla karşılaşılan diğer şikayetlerdir. Sıklıkla 20-30 yaşlarında rastlanmakla birlikte daha erken yaşlarda da görülebilir. Orta yaşlarda arka kutup çok sayıda yuvarlak, belirgin sınırlı beyaz renkte mozaik paternde yerleşen noktalar ile kaplanır. Sıklıkla iki gözde dikkat çekici şekilde bir simetri gözlenir. Genellikle foveaya yakın olanlar daha büyük izlenir. Dominant druzende bu benekler yuvarlak, beyaz ve fundus flavimakulatusa göre daha belirgin sınırlara sahiptir. İleri dönemlerde bu benekler özellikle santralde konfluen bir hal alır ve retina atrofik bir pigment epiteli görünümü kazanır. Daha sonra pigmentasyon meydana gelir ve koriokapillaris atrofisi ile geniş koroid damarların görünür hal alması izlenir. Nadiren druzen atrofik bir alan bırakarak kaybolabilir. Tipik özelliği bu druzenin YBMD nin aksine optik disk nazaline uzanım göstermesidir. Optik disk, damarlar ve retina periferi normal izlenir (55).
Resim 43: Her iki gözde simetrik yerleşimli, optik sinir nazaline de yayılım gösteren dominant druzen olgusu. Drusen yapılarının nazalde de olması dikkat çekicidir.
Kuzey Carolina Makula Distrofisi
Kuzey Carolina Makula distrofisi (KKMD), bilateral ve simetrik fundus bulguları ile seyreden otozomal dominant geçişli bir hastalıktır. Hayatın ilk 10 yılında makülada sarı noktalar ortaya çıkar ve zamanla koryoretinal atrofi, maküler stafilom ve periferik druzen gelişir. Atrofik makuler stafilom gelişene kadar santral görme normal düzeyde korunur. Görme keskinliği 1/10’un altına düşebilir (64).
Klinik spektrumu 3 evre olarak izlenir; evre 1: foveada druzen benzeri lezyonlar ve pigment değişiklikleri, evre 2: foveada birleşmiş druzen benzeri lezyonlar, evre 3: makulada koryoretinal atrofi ve stafilom görünümü (64).
Renkli görme korunur, görme alanında santral skotom vardır ve perifer korunmuştur. FA’de evre 1 ve 2’de druzen benzeri lezyonlar geç boyanır, evre 3’te koryokapillarisin nonperfüzyonu izlenir.
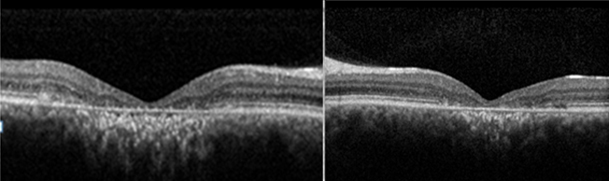
OKT’de evre 3’te sklerayı içermeyen koroidiçi derin çukurlaşmalar bulunur (Resim 44).
ERG ve EOG normaldir.
Resim 44: Muhtemel bir Kuzey Carolina Makula distrofisi olgumuzda (Evre 3) OKT’de sklerayı içermeyen koroid içi derin çukurlaşma görülmekte.
Koni Distrofisi
Çok sayıda koni disfonksiyonu bulunmaktadır. Bunlar progresif veya nonprogresif olabilir. Koni distrofisinde (KD), bütün Mendelian geçiş patenleri gösterilmiştir ancak otozomal resesif geçiş en sıktır. Koni fonksiyonu başlangıçta normal izlenir ancak hayatın ilk veya ikinci on yılında görme keskinliğinde azalma ve renkli görmede bozulma başlar. Fotofobi ve hemeralopi (gece körlüğü) eşlik edebilir. X’e bağlı geçen KD hastalarında hemeralopi ilk semptom olarak karşımıza çıkabilir, bu hastalarda tipik olarak 6 diyoptrinin üstünde miyopi saptanır.
KD’de koni fonksiyonu başta normal olduğu için nistagmus genellikle gözlenmez. Makulada sağlıklı görünümden, “hedef tahtası makulopati” ya da RPE atrofisine kadar çeşitli görünümde patolojiler izlenebilir (Resim 45). Optik diskte temporal solukluk bulunabilir. Görme alanında santral skotom varlığında periferik görme alanı korunmuştur. ERG’de azalmış kon cevabı varlığında rod cevabının korunmuş olması tanı açısından destekleyicidir, ancak hastalığın ilerleyen evrelerinde rodlarda da fonksiyon bozukluğu tabloya eklenebilir (65).
Genellikle adolesan dönemde semptomatik hale geçen hastalığın gelişimi çeşitlilik gösterse de görme keskinliği 50 yaşından önce legal körlük seviyesine kadar düşer.
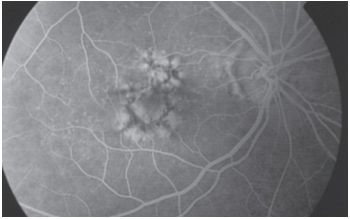
Resim 45: Koni distrofisi, bilateral santral atrofi ve FA da hedef tahtası görüntüsü
Santral (areolar) Koroidal Distrofi
Otozomal dominant geçişli bir hastalıktır, otozomal resesif vakalar da bildirilmiştir. Genç erişkinlerde makulada ince depigmentasyonla başlar. Zamanla keskin sınırlı simetrik retina ve koroid atrofisi gelişir. Görme keskinliği ve renkli görme azalır. Periferik görme alanı normaldir. Santral ve/veya parasantral skotom daima mevcuttur.
Floresein anjiografide pigment epiteli ve koriokapillaris atrofisine bağlı olarak koroid damarları izlenir.
Karanlık adaptasyonu genellikle normaldir. Bazı hastalarda hafifçe patolojik karanlık adaptasyon eğrileri görülebilir. Birçok hastada ERG normaldir. Ancak, özelikle santral koriokapillaris distrofisi, soluk diski ve incelmiş damarları olan olgularda subnormal değerler elde edilebilir (66).
HİPOTONİ MAKULOPATİSİ
Hipotoni makulopatisi ilk olarak Dellaporta tarafından 1954’te tanımlanmıştır . Hipotoni göz içi basıncının (GİB) 6 mmHg’nın altında olması, klinik olarak anlamlı hipotoni ise göz içi basıncının görme kaybına neden olacak kadar düşük olmasıdır. GİB düşüklüğüne bağlı görme kaybının nedenleri; hipoton makulopatisi, keratopati, katarakt, koroid efüzyonu, optik sinir ödemi, irregüler astigmatizma ve fitizis bulbidir (67).
Hipoton makulopatisi, en sık glokom cerrahisi ve penetran göz yaralanmasına ikincil meydana gelir. Travma, kronik üveitler, retina dekolmanı diğer etyolojik faktörlerdir. Hipotoniye ikincil gelişen papilödem, vasküler kıvrımlanma artışı ve koryoretinal katlantılar gibi fundus bulguları ile karakterizedir. Hipoton makulopatisinde skleral duvar yüzey gerilimini kaybeder, içe doğru kollabe olur, bu da koroid ve retinada kırışıklıkların oluşmasına neden olur. Vitreus kavitesinin ön-arka çapı azalır, kalın perifoveal retina çevrelediği ince foveal retinaya doğru sürüklenerek radial katlantılar oluşturur (68).
Katlantıların tepesi sarı renkli görülürken, çukur kısmı daha koyu bir görünüm vermektedir. FA koryoretinal katlantıları görüntülemede faydalıdır. Retina pigment epiteli seviyesinde alternan hiperfloresan ve hipofloresan çizgilenmeler gösterir. Tepe kısımlarında RPE incelmiş ve atrofiye uğramış olduğundan arka plandaki koroidal floresans artmıştır, çukur kısımlarda ise RPE arkaplan floresansını gizleyip hipofloresansa yol açar (68).
OKT, GİB’ı düşük ve görme kaybı bulunan hastalarda oftalmoskopi ve FA bulgusu yok ise tanı koydurucudur (Resim 46) (69).
Glokom filtrasyon cerrahilerinden sonra %20 oranında görüldüğü bildirilen hipoton makulopatisi sıklığı antimetabolitlerin kullanımı ile artmıştır. Genç yaş, miyopi, primer filtran cerrahi, sistemik hastalık varlığı ve preoperatif GİB’nın yüksek olması hipoton makulopatisi gelişimi için risk faktörleridir (67).
Ayırıcı tanıda koroidal katlantılara neden olabilen ve hipotoninin eşlik etmediği; hipermetropi, retrobulber kitle, arka sklerit, skleral çökertme, koroid tümörleri ve koroid neovaskülarizasyonu gözönünde bulundurulmalıdır.
Hipoton makulopatisinin tedavisi altta yatan nedene yönelik olmalıdır. Hedef, GİB’nı en kısa sürede normal seviyesine yükseltmektir. Gecikmiş müdahale kalıcı makuler koryoretinal değişikliklere ve görme kaybına neden olabilir (68).
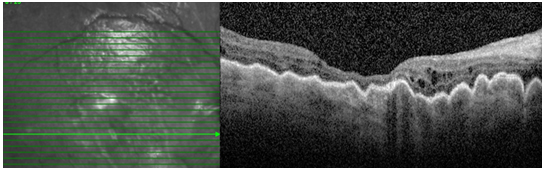
Resim 46: Hipotonide tipik koryoretinal katlantıların OKT deki görünümü.
TOKSİK MAKULOPATİLER
Klorokin ve Hidroksiklorokin
Klorokin ilk olarak malarya tedavisinde kullanılmıştır. Daha sonra romatoid artrit ve sistemik lupus eritematozus gibi kollajen doku hastalıklarının tedavisinde de kullanılmaya başlanmıştır. Günümüzde hidroksiklorokin kullanımı tercih edilmektedir. Klorokin ve hidroksiklorokin tedavi ve toksik dozları farklı olmakla beraber benzer makulopati tablosunu oluşturmaktadırlar (70).
Retinopati insidansı ilacın dozu ve kullanım süresi ile orantılı olarak artmaktadır. Sıklıkla kullanılan doz olan 400mg/gün doz ile 1000gr lık kümülatif doza 7 yılda ulaşılmaktadır. Bu nedenle 5-7 yıldan önce retinopati gelişmesi pek beklenmez. Hidroksiklorokin için 5 mg/kg/gün dozunda 10 yıl kullanımın düşük risk taşıdığı bildirilmiştir (70). Literatürde insidans %0-4 olarak bildirilmektedir. Wolke ve ark nın 4000 hastalık çalışmasında binde 6.8 olan retinal toxisite oranı, 5 yıldan daha uzun süredir kullananlar arasında birden artmakta ve %1 düzeyine çıkmaktadır (71,72). Fakat daha minyon yapılı kişilerde ve böbrek hastalığı olanlarda bu doz daha erken dönemde toksik doza ulaşabilmektedir.
Hidroksiklorokin kullanması planlanan hastaların öncelikle bir oftalmolojik muayeneden geçirilmeli ve fundus fotoğrafi (varsa) ve santral 10-2 görme alanına ilaveten en az bir objektif test [yüksek çözünürlüklü spektral OKT veya fundus otofloresans (FOF) veya multifokal ERG] yapılarak baseline durumları dokümante edilmeli ve bunlar bir referans noktası olarak kaydedilmelidir. Bu muayenede yaşa bağlı maküla dejenerasyonu gibi bazı maküla hastalıklarının erken dönem belirtileri varsa bu durum tedaviye başlamak için bir kontrendikasyon olarak değerlendirilmelidir. Eğer ilk muayenede bir sorun yok ve extra bir risk faktörü yoksa 5 yıldan önce yıllık taramalara başlamanın gereği yoktur (72).
Tablo 3: Hidroksiklorokin makulopatisi gelişiminde düşük-yüksek risk kriterleri (72)
| Hidroksiklorokin tedavisi | Düşük risk | Yüksek risk |
| Günlük doz | <6.5 mg/kg<200-400mg/gün | >6.5mg/kg,>400mg/gün |
| Kullanım süresi | <5 yıl | >5 yıl |
| Kümülatif doz | <1000gr(toplam) | >1000 gr(toplam) |
| Böbrek/karaciğer disfonksiyonu | – | + |
| Retina hastalığı ya da makulopati* | – | + |
| Yaş | İleri yaş |
*Retina hastalığı olan hastalar toksisite açısından yüksek riskli kabul edilir ve hidroksiklorokin kullanımı kontrendike kabul edilir çünkü toksisite gelişiminin erken belirtilerini maskeleyeceğinden tarama testlerini yanıltır.
Makulopati gelişen hastalar asemptomatik olabilir. Semptomatik hastalar okuma güçlüğü, renkli görmede bozulmalar veya santral, parasantral skotomlara bağlı gelişen ince becerilerde zorlanma şikayetleri ile başvururlar. Erken dönem bulgular görme alanında, FOF da, multifokal ERG ve OCTde kendini gösterebilir. İlk şüphe uyandıran belirtiler durumunda ilacın kesilmesi konusunda romatologlarla görüşülmesi ve ortak bir karar varılması gereklidir.
Fundus görünümü skotomlar geliştiği zaman dahi tamamen normal olabilir. Erken bulgular makulada pigment düzensizlikleri ve fovea reflesinin kaybolmasıdır. Zamanla merkezdeki pigment düzensizliğini hipopigmente bir alan çevreler. Bu parasantral hipopigmentasyon “Bull’s eye” (hedef tahtası) makulopatiye ilerler. İlaca maruziyet devam ederse pigment değişiklikleri yaygın bir hal alır ve periferde de pigment değişiklikleri görülür (70). Bu değişiklikler kalıcı olduğu gibi, ilaç kesildikten sonra dahi bir süre ilerleme göstermeye meyillidir. Bu kişiler stabilize olana kadar 3 ayda bir takip edilmelidirler.
Tamoksifen
Tamoksifen, bir selektif östrojen reseptör modülatörüdür, meme kanseri tedavisinde düşük dozlarda (20-40 mg/gün), kafa içi tümörlerin tedavisinde daha yüksek dozlarda (200 mg/gün) kullanılmaktadır. Hastaların 12%’sinde Tamoksifene bağlı gelişen oküler patolojiler bildirilmiştir. Bunlar; kristalin makulopatisi, kistoid maküler ödem, optik nörit ve korneal kristal depozitleridir (73).
Tamoksifen makulopatisi nadir görülen bir durumdur. Perifoveal alanda retina içi kristalin depozitlerinin birikmesi ile karakterizedir. Görme keskinliği genellikle foveal kistlerin gelişimine ikincil olarak düşer (74). Bu hastalar spektral OCT ile takip edilmeli ve makulopati belirtileri ilk saptandığında ilacın kullanımı sonlandırılmalıdır. Görme kaybının olmadığı asemptomatik olgularda retinopati tamoksifenin bırakılması ile geriler. KMÖ tedavisinde intravitreal anti-VEGF ajanlar ve triamsinolon kullanımı bildirilmiştir (73).
Tioridazin ve Klorpromazin
Tioridazin ve klorpromazin, şizofreni ve psikoz tedavisinde kullanılan antipsikotik ilaçlardır. Tioridazinin günlük dozu 150-600 mg’dır. 800 mg/gün üstünde tedavi ile birkaç hafta içinde görme keskinliğinde azalmaya neden olabilir. Fundus muayenesinde arka kutup ve perifer retinada “tuz-biber” manzarası olarak adlandırılan pigment değişiklikleri izlenir.
Klorpromazin günlük dozunun (75-300 mg/gün) üstünde uzun süre kullanıldığında retinada pigment değişiklikleri ile karakterize retinopati tablosuna neden olur (75).
Niasin (nikotinik asit, vit B3)
Niacin, hiperlipidemide destek tedavisi olarak kullanılır. Günde 1.5 gr üzerinde kullanımı ile bulanık görme, kapak ödemi, toksik ambliyopi, kirpik ve kaşlarda dökülme, yüzeyel punktat keratit, ve kistoid makula ödemi oküler yan etkilere neden olabilir. Kistoid makula ödemi görme keskinliğinde azalmaya neden olur, FFAda sızıntı izlenmez. Yan etkiler ilacın kesilmesi ile geriler (76).
Klofazimin
Klofazimin, kazanılmış immün yetmezliği olan hastalarda mikobakterium avium enfeksiyonlarının tedavisinde kullanılan antimikrobiyal bir ajandır. “Bull’s eye-hedef tahtası” makulopati ve yaygın retina dejenerasyonu yaptığı bildirilmiştir (77).
Fingolimod
Fingolimod bir yeni sınıf sfingozin-1-fosfat reseptör (S1FR) modülatörüdür. Tekrarlayan atakları olan multiple skleroz (MS) hastalarında günde 0.5 mg dozunda oral kullanımı onaylanmıştır. Fingolimod’un 0.5 mg/gün dozunda kullanımının makula ödemine yol açabileceği gösterilmiştir (78). Fingolimod tedavisine başlamadan önce hastalara göz muayenesi yapılması ve tedavi süresince düzenli takiplerin devam ettirilmesi önerilmektedir.